

Epidemiological and molecular study of hemoglobinopathies in Mauritanian patients
- PMID: 36106931
- PMCID: PMC9544207
- DOI: 10.1002/mgg3.2048
Epidemiological and molecular study of hemoglobinopathies in Mauritanian patients
Abstract
Background: Hemoglobinopathies, inherited disorders of hemoglobin (Hb), are the most common hereditary monogenic diseases of the red cell in the world. Few studies have been conducted on hemoglobinopathies in Mauritania. Therefore, the aim of this work is to establish the molecular and epidemiological basis of hemoglobinopathies in a cohort of Mauritanian patients and to determine the haplotype of the β-globin gene cluster in sickle cell subjects.
Methods: The molecular screening of Hb disorders in 40 Mauritanian patients was done by a polymerase-restriction fragment length polymorphism (RFLP) for the sickle cell disease (SCD) mutation, a PCR/sequencing method for β-thalassemia mutations, and by the multiplex polymerase chain reaction method for the α-thalassemia. The exploration of eight polymorphic sites (SNPs) within the β-globin gene cluster was conducted by PCR/RFLP method, to identify the HbS haplotypes from the sickle cell subjects.
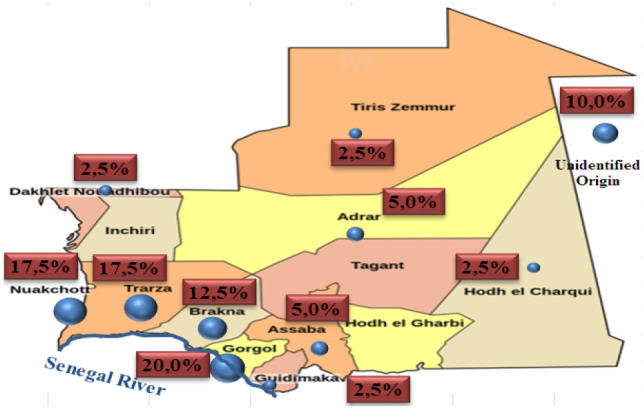
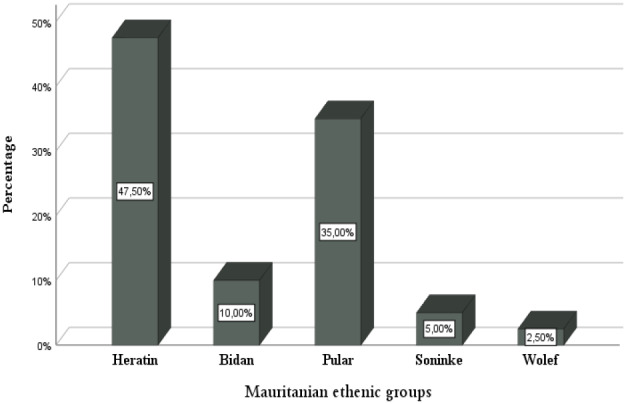
Results: The epidemiological study of our patients showed a high incidence in the Senegal River area (52.5%) and a high ethnic prevalence for the Heratin (47.5%) and the Pular (35%). Molecular study allowed us to identify eight different mutations in our sample analyzed. They are respectively: HbS (HBB:c.20A>T) (68.75%), Cd44 -C (HBB:c.135delC) (8.75%), -29A>G (HBB:c.-79A>G) (4.8%), -α-3.7 (g.34164_37967del3804) (3.75%), IVS-II-849A>G (HBB:c.316-2A>G) (2.25%) and Cd24T>A (HBB:c.75T>A), Hb Siirt (HBB:c.83C>G) and HbC (HBB:c.19G>A) each with (1.25%). Six different haplotypes are being explored among the SCD subjects with the Senegal haplotype as the most prevalent (66.7%), followed by Benin (10%), Arab-Indians (6.7%), Bantu (3.3%), and two atypical haplotypes.
Conclusion: Our findings enrich the epidemiological data in our population and could contribute to the establishment of a strategy of prevention and management through screening, genetic counseling, and prenatal diagnosis of Hemoglobinopathies in the Mauritanian population.
Keywords: haplotypes; hemoglobin (Hb) disorders; hemoglobinopathies; sickle cell disease (SCD); α-thalassemia; β-thalassemia.
© 2022 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals LLC.
Conflict of interest statement
The authors report no conflict of interest.
Figures

Similar articles
-
Profiling of 35 Cases of Hb S/Hb E (HBB: c.20A>T/HBB: c.79G>a), Disease and Association with α-Thalassemia and β-Globin Gene Cluster Haplotypes from Odisha, India.Hemoglobin. 2021 Nov;45(6):380-386. doi: 10.1080/03630269.2021.1965618. Epub 2022 Mar 4. Hemoglobin. 2021. PMID: 35243949
-
Unusual β-Globin Haplotype Distribution in Newborns from Bengo, Angola.Hemoglobin. 2019 May;43(3):149-154. doi: 10.1080/03630269.2019.1647230. Epub 2019 Aug 8. Hemoglobin. 2019. PMID: 31394941
-
Ten Years of Routine α- and β-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations.Hemoglobin. 2016;40(2):75-84. doi: 10.3109/03630269.2015.1113990. Epub 2015 Dec 4. Hemoglobin. 2016. PMID: 26635043 Review.
-
Chronic inflammatory state in sickle cell anemia patients is associated with HBB(*)S haplotype.Cytokine. 2014 Feb;65(2):217-21. doi: 10.1016/j.cyto.2013.10.009. Epub 2013 Nov 27. Cytokine. 2014. PMID: 24290434
-
Beta-globin gene haplotypes among cameroonians and review of the global distribution: is there a case for a single sickle mutation origin in Africa?OMICS. 2015 Mar;19(3):171-9. doi: 10.1089/omi.2014.0134. OMICS. 2015. PMID: 25748438 Free PMC article. Review.
References
-
- Amselem, S. , Nunes, V. , Vidaud, M. , Estivill, X. , Wong, C. , d'Auriol, L. , Vidaud, D. , Galibert, F. , Baiget, M. , Goossens, M. , Amselem, S. N. V. , & Vidaud, M. (1988). Determination of the spectrum of β thalassemia genes in Spain by use of dot blot analysis of amplified β globin DNA. American Journal of Human Genetics, 43, 95–100. - PMC - PubMed
-
- Antonarakis, S. E. , Irkin, S. H. , Cheng, T. C. , Scott, A. F. , Sexton, J. P. , Trusko, S. P. , Charache, S. , & Kazazian, H. H., Jr. (1984). β‐Thalassemia in American blacks: Novel mutations in the “TATA” box and an acceptor splice sites. Proceedings of the National Academy of Sciences of the United States of America, 81(4), 1154–1158. - PMC - PubMed
-
- Arnold, S. C. , Quah, T. C. , Low, P. S. , & Chong, S. S. (2001). A rapid and reliable 7‐deletion multiplex polymerase chain reaction assay for a‐thalassemia. Blood, 98(1), 250–251. - PubMed
-
- Boudrahem‐Addour, N. , Zidani, N. , Carion, N. , Labie, D. , Belhani, M. , & Beldjord, C. (2009). Molecular heterogeneity of beta‐thalassemia in Algeria: How to face up to a major health problem. Hemoglobin, 33, 34–36. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
