

Benchmark datasets for SARS-CoV-2 surveillance bioinformatics
- PMID: 36093336
- PMCID: PMC9454940
- DOI: 10.7717/peerj.13821
Benchmark datasets for SARS-CoV-2 surveillance bioinformatics
Abstract
Background: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the cause of coronavirus disease 2019 (COVID-19), has spread globally and is being surveilled with an international genome sequencing effort. Surveillance consists of sample acquisition, library preparation, and whole genome sequencing. This has necessitated a classification scheme detailing Variants of Concern (VOC) and Variants of Interest (VOI), and the rapid expansion of bioinformatics tools for sequence analysis. These bioinformatic tools are means for major actionable results: maintaining quality assurance and checks, defining population structure, performing genomic epidemiology, and inferring lineage to allow reliable and actionable identification and classification. Additionally, the pandemic has required public health laboratories to reach high throughput proficiency in sequencing library preparation and downstream data analysis rapidly. However, both processes can be limited by a lack of a standardized sequence dataset.
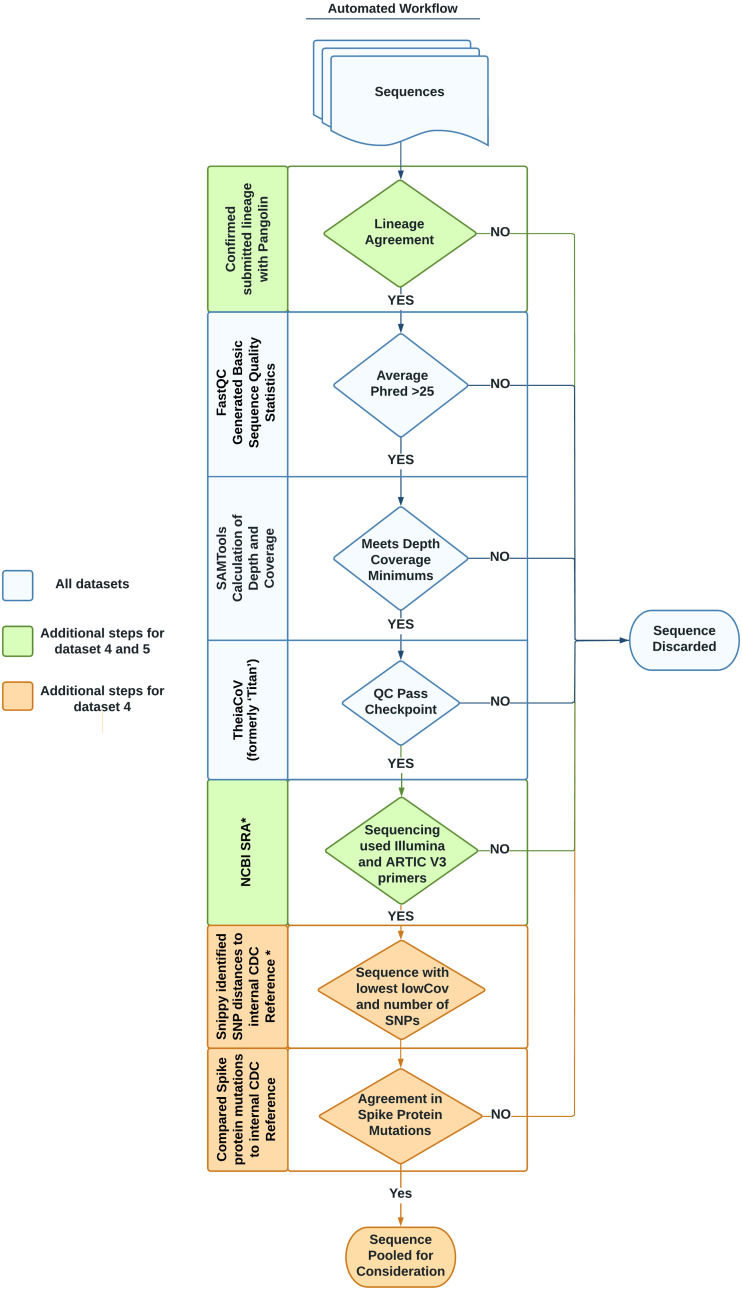
Methods: We identified six SARS-CoV-2 sequence datasets from recent publications, public databases and internal resources. In addition, we created a method to mine public databases to identify representative genomes for these datasets. Using this novel method, we identified several genomes as either VOI/VOC representatives or non-VOI/VOC representatives. To describe each dataset, we utilized a previously published datasets format, which describes accession information and whole dataset information. Additionally, a script from the same publication has been enhanced to download and verify all data from this study.
Results: The benchmark datasets focus on the two most widely used sequencing platforms: long read sequencing data from the Oxford Nanopore Technologies platform and short read sequencing data from the Illumina platform. There are six datasets: three were derived from recent publications; two were derived from data mining public databases to answer common questions not covered by published datasets; one unique dataset representing common sequence failures was obtained by rigorously scrutinizing data that did not pass quality checks. The dataset summary table, data mining script and quality control (QC) values for all sequence data are publicly available on GitHub: https://github.com/CDCgov/datasets-sars-cov-2.
Discussion: The datasets presented here were generated to help public health laboratories build sequencing and bioinformatics capacity, benchmark different workflows and pipelines, and calibrate QC thresholds to ensure sequencing quality. Together, improvements in these areas support accurate and timely outbreak investigation and surveillance, providing actionable data for pandemic management. Furthermore, these publicly available and standardized benchmark data will facilitate the development and adjudication of new pipelines.
Keywords: Benchmarking; COVID-19; Standardization; WGS; sha256.
©2022 Xiaoli et al.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
Similar articles
-
Unlocking data: Decision-maker perspectives on cross-sectoral data sharing and linkage as part of a whole-systems approach to public health policy and practice.Public Health Res (Southampt). 2024 Nov 20:1-30. doi: 10.3310/KYTW2173. Online ahead of print. Public Health Res (Southampt). 2024. PMID: 39582242
-
Falls prevention interventions for community-dwelling older adults: systematic review and meta-analysis of benefits, harms, and patient values and preferences.Syst Rev. 2024 Nov 26;13(1):289. doi: 10.1186/s13643-024-02681-3. Syst Rev. 2024. PMID: 39593159 Free PMC article.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Trends in Surgical and Nonsurgical Aesthetic Procedures: A 14-Year Analysis of the International Society of Aesthetic Plastic Surgery-ISAPS.Aesthetic Plast Surg. 2024 Oct;48(20):4217-4227. doi: 10.1007/s00266-024-04260-2. Epub 2024 Aug 5. Aesthetic Plast Surg. 2024. PMID: 39103642 Review.
Cited by
-
Genome-wide identification and molecular evolution of elongation family of very long chain fatty acids proteins in Cyrtotrachelus buqueti.BMC Genomics. 2024 Aug 2;25(1):758. doi: 10.1186/s12864-024-10658-8. BMC Genomics. 2024. PMID: 39095734 Free PMC article.
-
Lessons learned: overcoming common challenges in reconstructing the SARS-CoV-2 genome from short-read sequencing data via CoVpipe2.F1000Res. 2024 Apr 16;12:1091. doi: 10.12688/f1000research.136683.1. eCollection 2023. F1000Res. 2024. PMID: 38716230 Free PMC article.
-
PHA4GE quality control contextual data tags: standardized annotations for sharing public health sequence datasets with known quality issues to facilitate testing and training.Microb Genom. 2024 Jun;10(6):001260. doi: 10.1099/mgen.0.001260. Microb Genom. 2024. PMID: 38860884 Free PMC article.
-
Bioinformatic investigation of discordant sequence data for SARS-CoV-2: insights for robust genomic analysis during pandemic surveillance.Microb Genom. 2023 Nov;9(11):001146. doi: 10.1099/mgen.0.001146. Microb Genom. 2023. PMID: 38019123 Free PMC article.
References
-
- Andrews S. Babraham bioinformatics—FastQC a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [03 November 2021];2010
-
- ARTIC Home—artic pipeline. 2020. https://artic.readthedocs.io/en/latest/?badgelatest. [30 November 2021]. https://artic.readthedocs.io/en/latest/?badgelatest
-
- Baker DJ, Aydin A, Le-Viet T, Kay GL, Rudder S, De Oliveira Martins L, Tedim AP, Kolyva A, Diaz M, Alikhan N-F, Meadows L, Bell A, Gutierrez AV, Trotter AJ, Thomson NM, Gilroy R, Griffith L, Adriaenssens EM, Stanley R, Charles IG, Elumogo N, Wain J, Prakash R, Meader E, Mather AE, Webber MA, Dervisevic S, Page AJ, O’Grady J. CoronaHiT: high-throughput sequencing of SARS-CoV-2 genomes. Genome Medicine. 2021;13:21. doi: 10.1186/s13073-021-00839-5. - DOI - PMC - PubMed
-
- BBMap https://sourceforge.net/projects/bbmap/ [03 November 2021];2021
Publication types
MeSH terms
Grants and funding
- U19 AI110818/AI/NIAID NIH HHS/United States
- BBS/E/F/000PR10352/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/CCG1860/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/R012504/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
