

Pharmacological suppression of Nedd4-2 rescues the reduction of Kv11.1 channels in pathological cardiac hypertrophy
- PMID: 36059970
- PMCID: PMC9428276
- DOI: 10.3389/fphar.2022.942769
Pharmacological suppression of Nedd4-2 rescues the reduction of Kv11.1 channels in pathological cardiac hypertrophy
Abstract
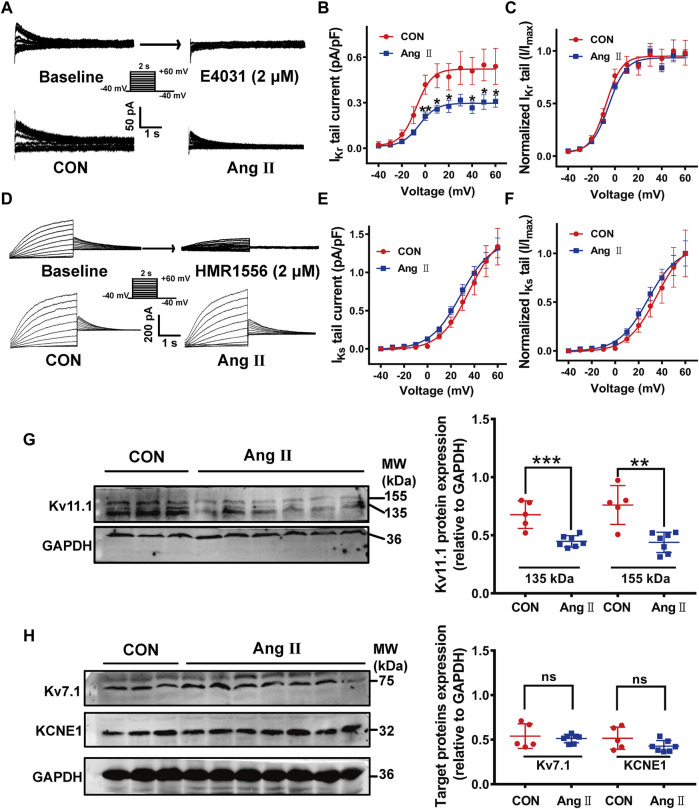
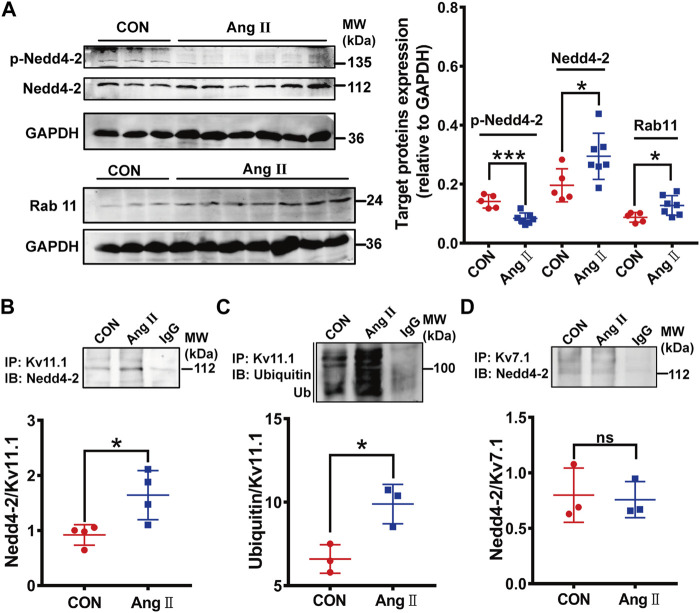
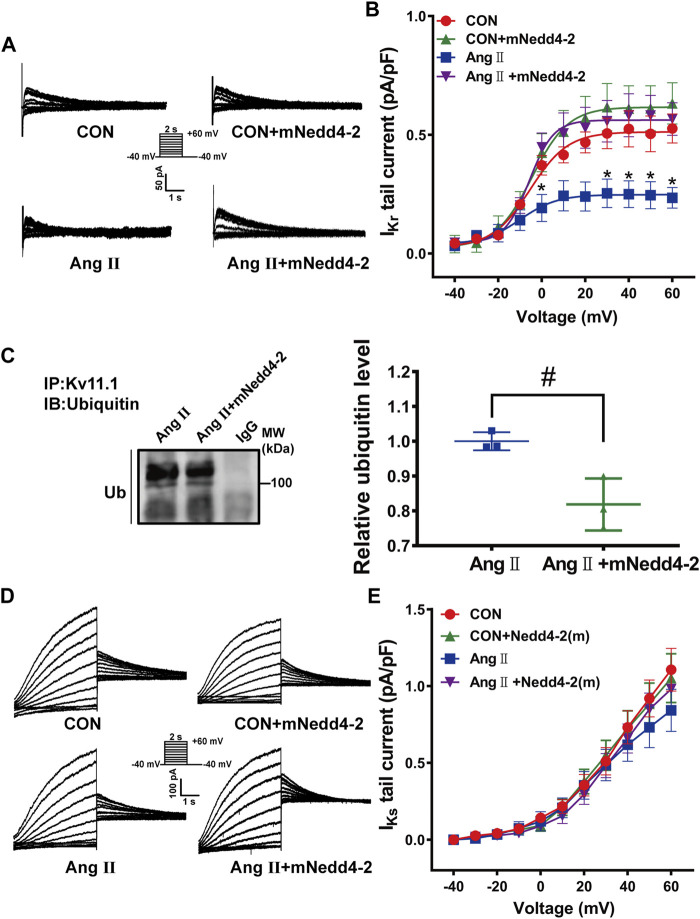
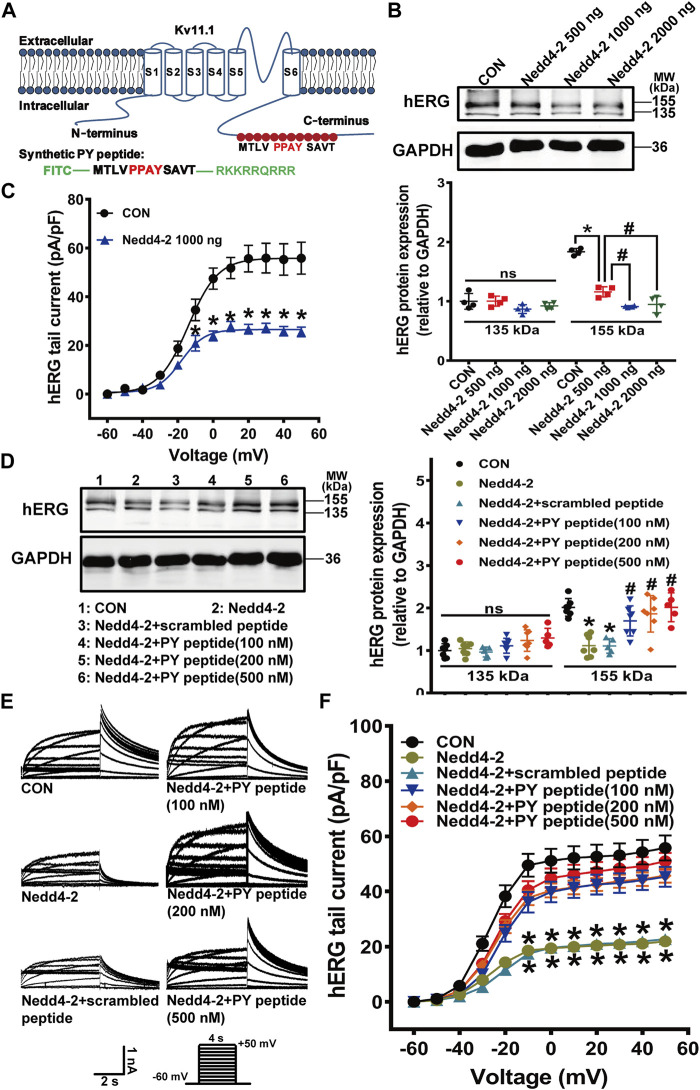
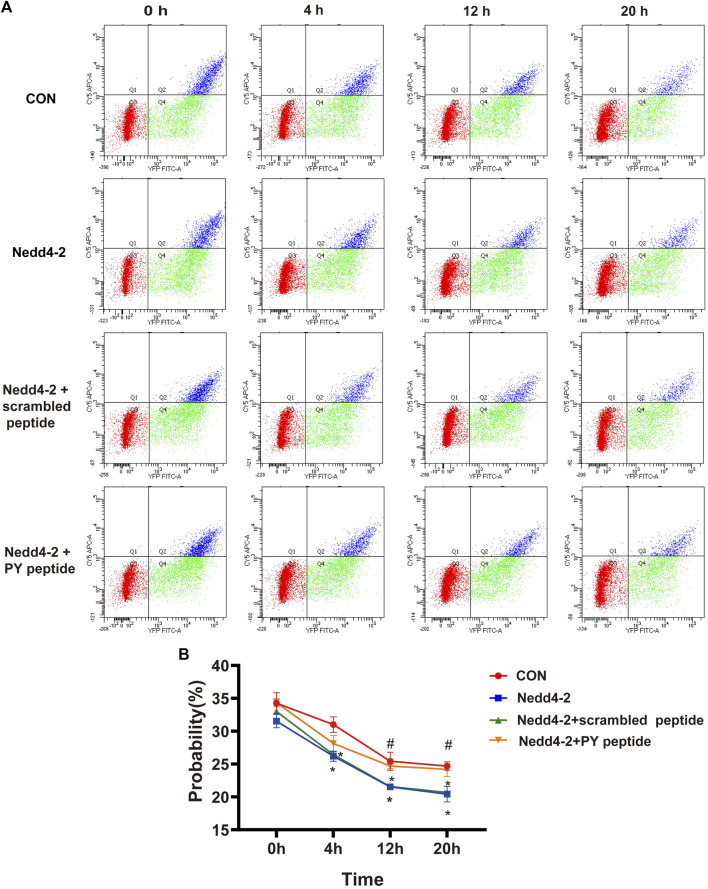
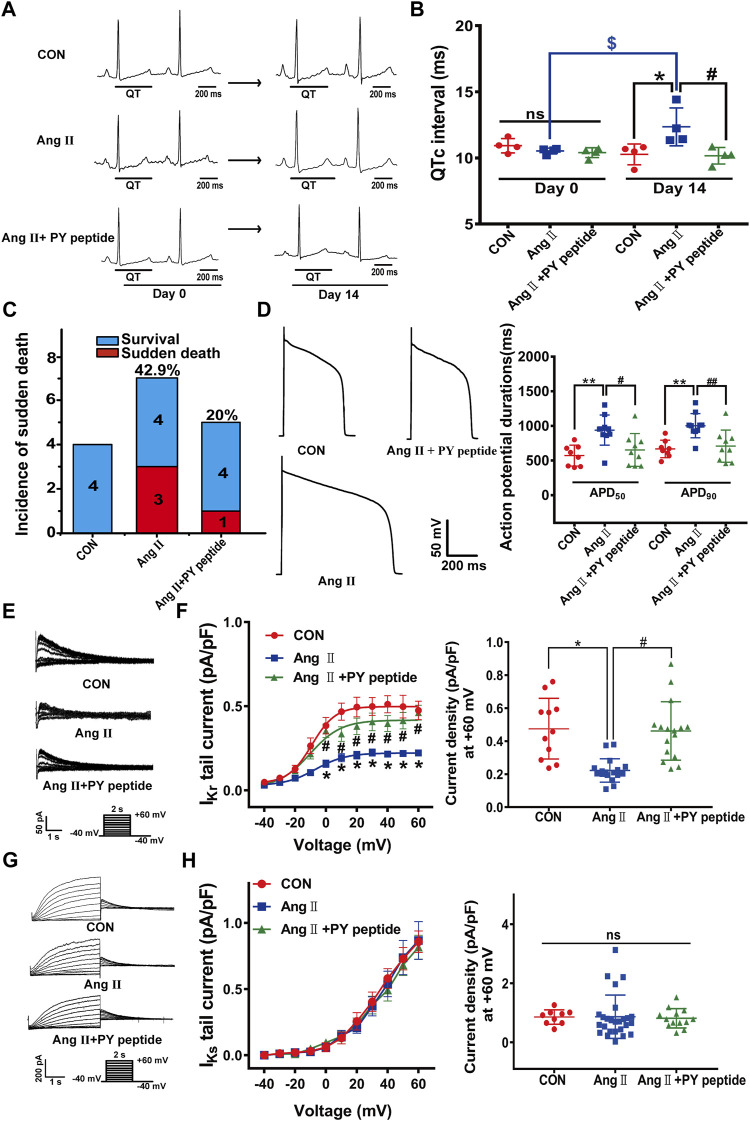
The human ether-á-go-go-related gene (hERG) encodes the pore-forming subunit (Kv11.1), conducting a rapidly delayed rectifier K+ current (I Kr). Reduction of I Kr in pathological cardiac hypertrophy (pCH) contributes to increased susceptibility to arrhythmias. However, practical approaches to prevent I Kr deficiency are lacking. Our study investigated the involvement of ubiquitin ligase Nedd4-2-dependent ubiquitination in I Kr reduction and sought an intervening approach in pCH. Angiotensin II (Ang II) induced a pCH phenotype in guinea pig, accompanied by increased incidences of sudden death and higher susceptibility to arrhythmias. Patch-clamp recordings revealed a significant I Kr reduction in pCH cardiomyocytes. Kv11.1 protein expression was decreased whereas its mRNA level did not change. In addition, Nedd4-2 protein expression was increased in pCH, accompanied by an enhanced Nedd4-2 and Kv11.1 binding detected by immunoprecipitation analysis. Cardiac-specific overexpression of inactive form of Nedd4-2 shortened the prolonged QT interval, reversed I Kr reduction, and decreased susceptibility to arrhythmias. A synthesized peptide containing the PY motif in Kv11.1 C-terminus binding to Nedd4-2 and a cell-penetrating sequence antagonized Nedd4-2-dependent degradation of the channel and increased the surface abundance and function of hERG channel in HEK cells. In addition, in vivo administration of the PY peptide shortened QT interval and action potential duration, and enhanced I Kr in pCH. We conclude that Nedd4-2-dependent ubiquitination is critically involved in I Kr deficiency in pCH. Pharmacological suppression of Nedd4-2 represents a novel approach for antiarrhythmic therapy in pCH.
Keywords: Nedd4-2; arrhythmia; pathological cardiac hypertrophy; rapid delayed rectifier K+ current; ubiquitination.
Copyright © 2022 Zhang, Fu, Sun, Zou, Qiu, Zhang, Su, Shi, Li and Xu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Regulation of the human ether-a-go-go-related gene (hERG) channel by Rab4 protein through neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2).J Biol Chem. 2013 Jul 26;288(30):21876-86. doi: 10.1074/jbc.M113.461715. Epub 2013 Jun 21. J Biol Chem. 2013. PMID: 23792956 Free PMC article.
-
Cell surface expression of human ether-a-go-go-related gene (hERG) channels is regulated by caveolin-3 protein via the ubiquitin ligase Nedd4-2.J Biol Chem. 2012 Sep 28;287(40):33132-41. doi: 10.1074/jbc.M112.389643. Epub 2012 Aug 9. J Biol Chem. 2012. PMID: 22879586 Free PMC article.
-
Regulation of the human ether-a-go-go-related gene (hERG) potassium channel by Nedd4 family interacting proteins (Ndfips).Biochem J. 2015 Nov 15;472(1):71-82. doi: 10.1042/BJ20141282. Epub 2015 Sep 11. Biochem J. 2015. PMID: 26363003
-
[HERG K+ channel, the target of anti-arrhythmias drugs].Yao Xue Xue Bao. 2007 Jul;42(7):687-91. Yao Xue Xue Bao. 2007. PMID: 17882949 Review. Chinese.
-
I(Kr): the hERG channel.J Mol Cell Cardiol. 2001 May;33(5):835-49. doi: 10.1006/jmcc.2000.1317. J Mol Cell Cardiol. 2001. PMID: 11343409 Review.
Cited by
-
Quercetin is a foe in the heart by targeting the hERG potassium channel.Iran J Basic Med Sci. 2024;27(11):1397-1404. doi: 10.22038/ijbms.2024.77846.16848. Iran J Basic Med Sci. 2024. PMID: 39386239 Free PMC article.
References
-
- Andersen M. N., Hefting L. L., Steffensen A. B., Schmitt N., Olesen S. P., Olsen J. V., et al. (2015). Protein kinase A stimulates Kv7.1 surface expression by regulating Nedd4-2-dependent endocytic trafficking. Am. J. Physiol. Cell. Physiol. 309 (10), C693–C706. 10.1152/ajpcell.00383.2014 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous