

A comprehensive comparison of multilocus association methods with summary statistics in genome-wide association studies
- PMID: 36042399
- PMCID: PMC9429742
- DOI: 10.1186/s12859-022-04897-3
A comprehensive comparison of multilocus association methods with summary statistics in genome-wide association studies
Abstract
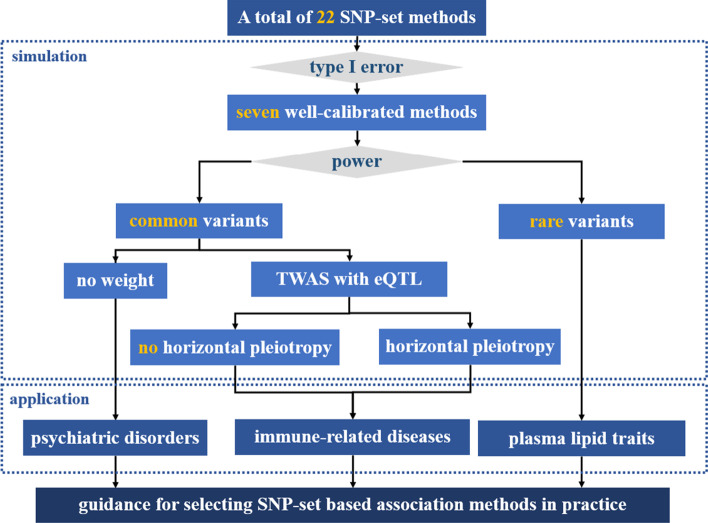
Background: Multilocus analysis on a set of single nucleotide polymorphisms (SNPs) pre-assigned within a gene constitutes a valuable complement to single-marker analysis by aggregating data on complex traits in a biologically meaningful way. However, despite the existence of a wide variety of SNP-set methods, few comprehensive comparison studies have been previously performed to evaluate the effectiveness of these methods.
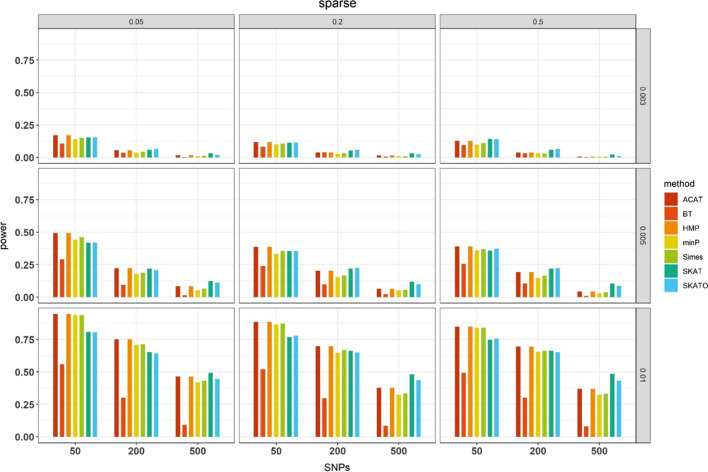
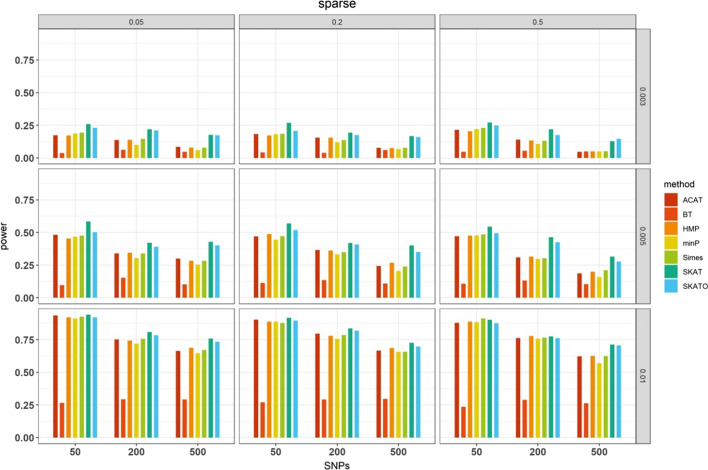
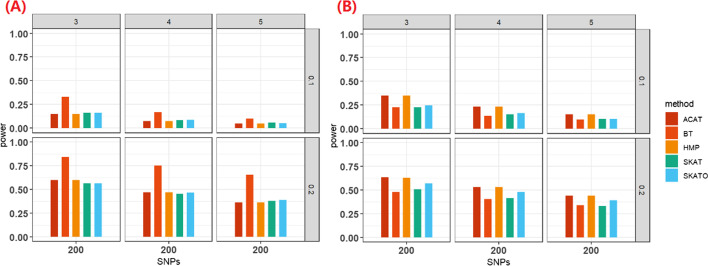
Results: We herein sought to fill this knowledge gap by conducting a comprehensive empirical comparison for 22 commonly-used summary-statistics based SNP-set methods. We showed that only seven methods could effectively control the type I error, and that these well-calibrated approaches had varying power performance under the simulation scenarios. Overall, we confirmed that the burden test was generally underpowered and score-based variance component tests (e.g., sequence kernel association test) were much powerful under the polygenic genetic architecture in both common and rare variant association analyses. We further revealed that two linkage-disequilibrium-free P value combination methods (e.g., harmonic mean P value method and aggregated Cauchy association test) behaved very well under the sparse genetic architecture in simulations and real-data applications to common and rare variant association analyses as well as in expression quantitative trait loci weighted integrative analysis. We also assessed the scalability of these approaches by recording computational time and found that all these methods can be scalable to biobank-scale data although some might be relatively slow.
Conclusion: In conclusion, we hope that our findings can offer an important guidance on how to choose appropriate multilocus association analysis methods in post-GWAS era. All the SNP-set methods are implemented in the R package called MCA, which is freely available at https://github.com/biostatpzeng/ .
Keywords: Common and rare variant association study; Expression quantitative trait loci; Genome-wide association study; Integrative analysis; Multilocus method; P value combination method; SNP-set analysis; Summary statistics.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
A gene based combination test using GWAS summary data.BMC Bioinformatics. 2023 Jan 3;24(1):2. doi: 10.1186/s12859-022-05114-x. BMC Bioinformatics. 2023. PMID: 36597047 Free PMC article.
-
Estimating colocalization probability from limited summary statistics.BMC Bioinformatics. 2021 May 17;22(1):254. doi: 10.1186/s12859-021-04170-z. BMC Bioinformatics. 2021. PMID: 34000989 Free PMC article.
-
SumVg: Total Heritability Explained by All Variants in Genome-Wide Association Studies Based on Summary Statistics with Standard Error Estimates.Int J Mol Sci. 2024 Jan 22;25(2):1347. doi: 10.3390/ijms25021347. Int J Mol Sci. 2024. PMID: 38279346 Free PMC article.
-
Comparison of methods for estimating genetic correlation between complex traits using GWAS summary statistics.Brief Bioinform. 2021 Sep 2;22(5):bbaa442. doi: 10.1093/bib/bbaa442. Brief Bioinform. 2021. PMID: 33497438 Free PMC article.
-
How powerful are summary-based methods for identifying expression-trait associations under different genetic architectures?Pac Symp Biocomput. 2018;23:228-239. Pac Symp Biocomput. 2018. PMID: 29218884 Free PMC article.
Cited by
-
The goldmine of GWAS summary statistics: a systematic review of methods and tools.BioData Min. 2024 Sep 5;17(1):31. doi: 10.1186/s13040-024-00385-x. BioData Min. 2024. PMID: 39238044 Free PMC article.
-
Detecting associated genes for complex traits shared across East Asian and European populations under the framework of composite null hypothesis testing.J Transl Med. 2022 Sep 23;20(1):424. doi: 10.1186/s12967-022-03637-8. J Transl Med. 2022. PMID: 36138484 Free PMC article.
-
Incorporating genetic similarity of auxiliary samples into eGene identification under the transfer learning framework.J Transl Med. 2024 Mar 9;22(1):258. doi: 10.1186/s12967-024-05053-6. J Transl Med. 2024. PMID: 38461317 Free PMC article.
References
-
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012. doi: 10.1093/nar/gky1120. - DOI - PMC - PubMed
MeSH terms
Grants and funding
- 2014LY112/the Statistical Science Research Project from National Bureau of Statistics of China
- 2018M630607/the China Postdoctoral Science Foundation
- BK20181472/the Natural Science Foundation of Jiangsu Province of China
- TD202008/the Training Project for Youth Teams of Science and Technology Innovation at Xuzhou Medical University
- WSN-087/the Six-Talent Peaks Project in Jiangsu Province of China
- KC20062/the Social Development Project of Xuzhou City
- WT_/Wellcome Trust/United Kingdom
- 82173630/National Natural Science Foundation of China
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- 18YJC910002/the Youth Foundation of Humanity and Social Science funded by Ministry of Education of China
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Miscellaneous
