

Processing DNA lesions during mitosis to prevent genomic instability
- PMID: 36040211
- PMCID: PMC9444068
- DOI: 10.1042/BST20220049
Processing DNA lesions during mitosis to prevent genomic instability
Abstract
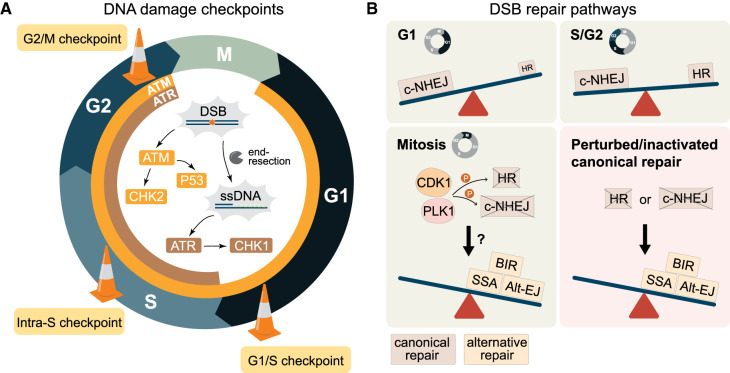
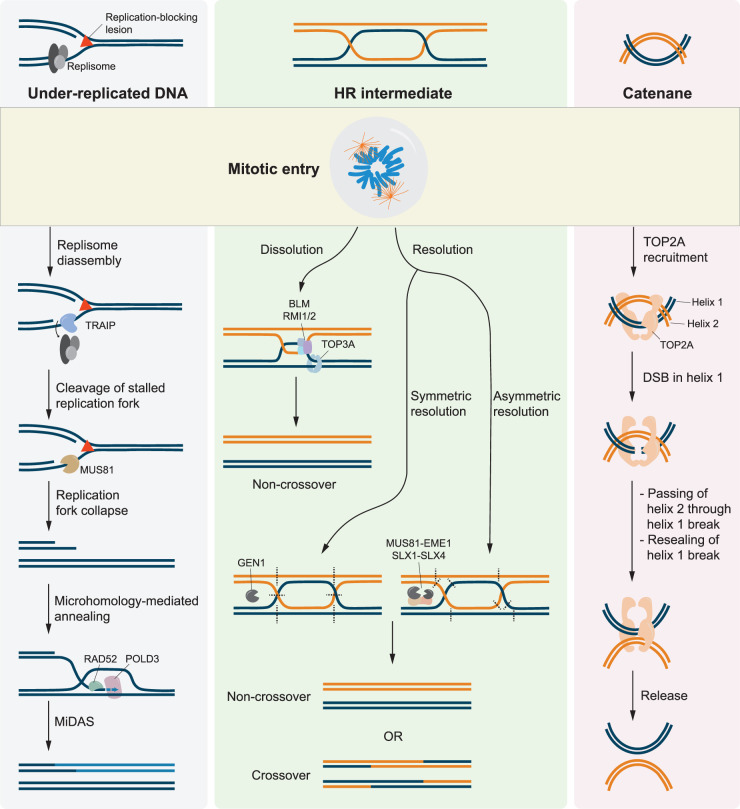
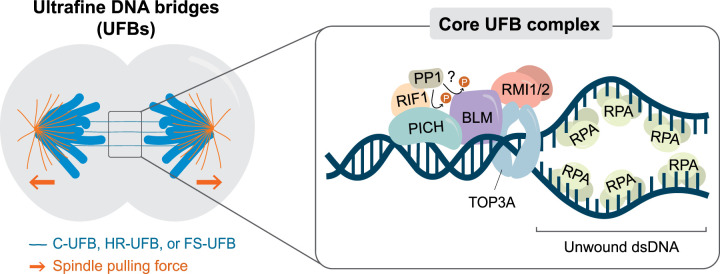
Failure of cells to process toxic double-strand breaks (DSBs) constitutes a major intrinsic source of genome instability, a hallmark of cancer. In contrast with interphase of the cell cycle, canonical repair pathways in response to DSBs are inactivated in mitosis. Although cell cycle checkpoints prevent transmission of DNA lesions into mitosis under physiological condition, cancer cells frequently display mitotic DNA lesions. In this review, we aim to provide an overview of how mitotic cells process lesions that escape checkpoint surveillance. We outline mechanisms that regulate the mitotic DNA damage response and the different types of lesions that are carried over to mitosis, with a focus on joint DNA molecules arising from under-replication and persistent recombination intermediates, as well as DNA catenanes. Additionally, we discuss the processing pathways that resolve each of these lesions in mitosis. Finally, we address the acute and long-term consequences of unresolved mitotic lesions on cellular fate and genome stability.
Keywords: DNA damage response; cell cycle; genome instability; genome integrity; mitosis.
© 2022 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures

Similar articles
-
Double-strand break repair-adox: Restoration of suppressed double-strand break repair during mitosis induces genomic instability.Cancer Sci. 2014 Dec;105(12):1519-25. doi: 10.1111/cas.12551. Epub 2014 Nov 5. Cancer Sci. 2014. PMID: 25287622 Free PMC article. Review.
-
How Cells Respond to DNA Breaks in Mitosis.Trends Biochem Sci. 2020 Apr;45(4):321-331. doi: 10.1016/j.tibs.2019.12.010. Epub 2020 Jan 27. Trends Biochem Sci. 2020. PMID: 32001093 Review.
-
Polθ is phosphorylated by PLK1 to repair double-strand breaks in mitosis.Nature. 2023 Sep;621(7978):415-422. doi: 10.1038/s41586-023-06506-6. Epub 2023 Sep 6. Nature. 2023. PMID: 37674080 Free PMC article.
-
Working on Genomic Stability: From the S-Phase to Mitosis.Genes (Basel). 2020 Feb 20;11(2):225. doi: 10.3390/genes11020225. Genes (Basel). 2020. PMID: 32093406 Free PMC article. Review.
-
Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events.Cell Cycle. 2011 Apr 15;10(8):1215-21. doi: 10.4161/cc.10.8.15334. Epub 2011 Apr 15. Cell Cycle. 2011. PMID: 21412056 Free PMC article.
Cited by
-
Improving Localized Radiotherapy for Glioblastoma via Small Molecule Inhibition of KIF11.Cancers (Basel). 2023 Jun 13;15(12):3173. doi: 10.3390/cancers15123173. Cancers (Basel). 2023. PMID: 37370783 Free PMC article.
-
The Influence of Edaphic Factors on DNA Damage and Repair in Wild Wheat Triticum dicoccoides Körn. (Poaceae, Triticeae).Int J Mol Sci. 2023 Apr 6;24(7):6847. doi: 10.3390/ijms24076847. Int J Mol Sci. 2023. PMID: 37047823 Free PMC article.
-
KAT5 histone acetyltransferase mutations in cancer cells.MicroPubl Biol. 2022 Nov 28;2022:10.17912/micropub.biology.000676. doi: 10.17912/micropub.biology.000676. eCollection 2022. MicroPubl Biol. 2022. PMID: 36530474 Free PMC article.
-
Msc1 is a nuclear envelope protein that reinforces DNA repair in late mitosis.iScience. 2024 Jun 11;27(7):110250. doi: 10.1016/j.isci.2024.110250. eCollection 2024 Jul 19. iScience. 2024. PMID: 39021806 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources