

Simple and accurate transcriptional start site identification using Smar2C2 and examination of conserved promoter features
- PMID: 36030508
- PMCID: PMC9827901
- DOI: 10.1111/tpj.15957
Simple and accurate transcriptional start site identification using Smar2C2 and examination of conserved promoter features
Abstract
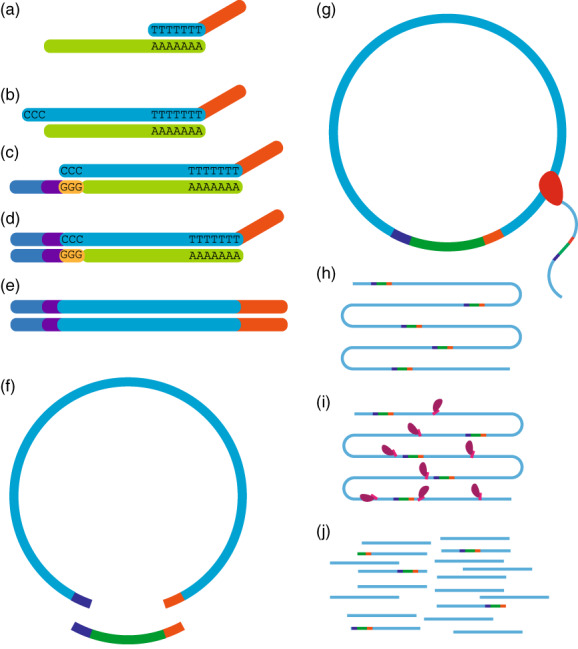
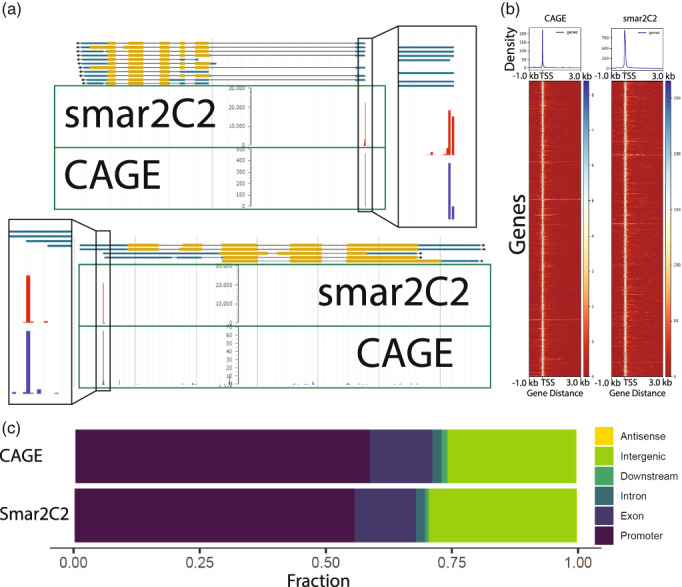
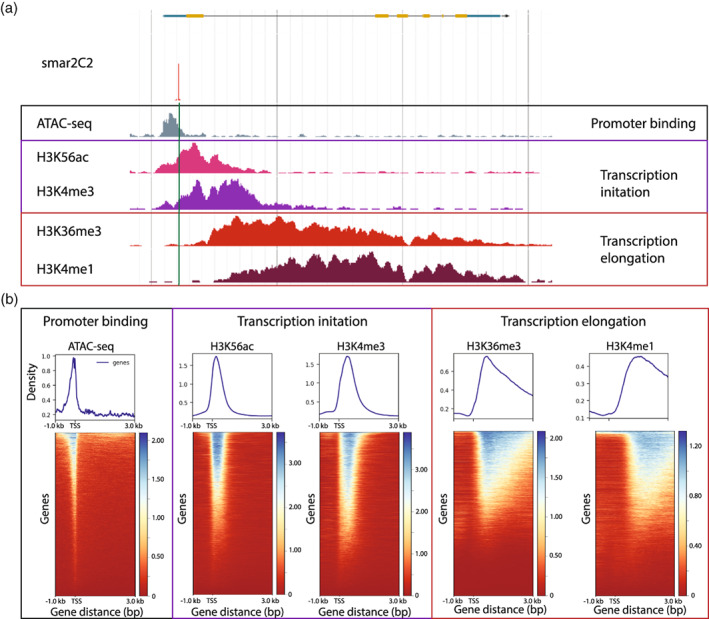
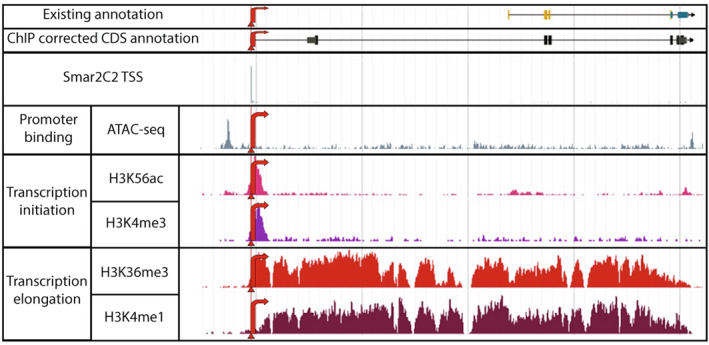
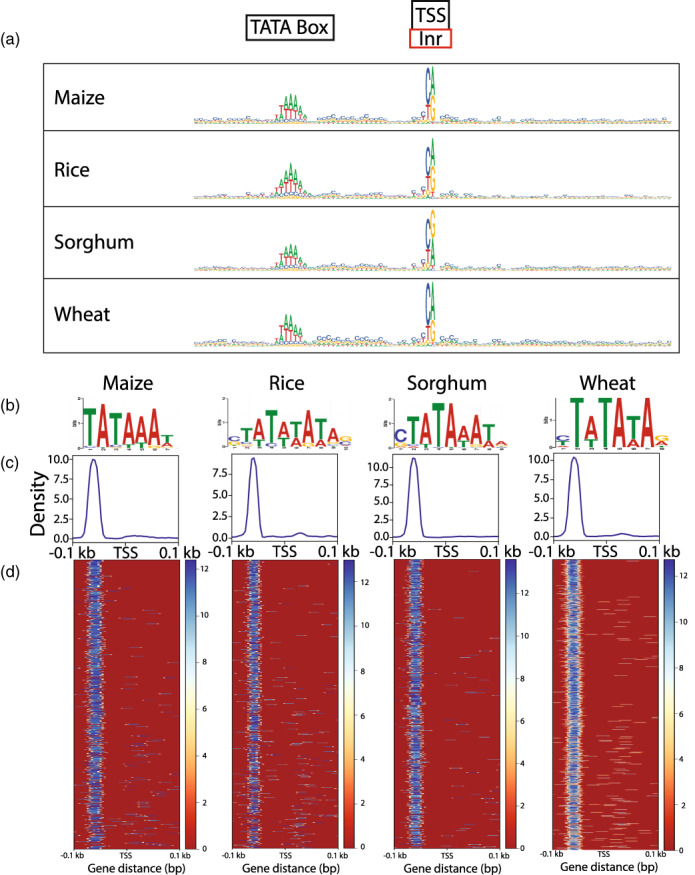
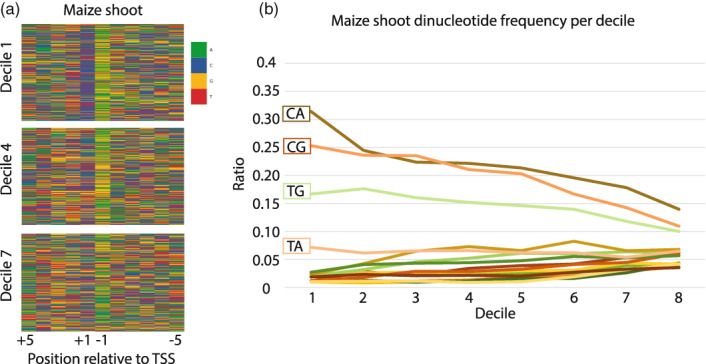
The precise and accurate identification and quantification of transcriptional start sites (TSSs) is key to understanding the control of transcription. The core promoter consists of the TSS and proximal non-coding sequences, which are critical in transcriptional regulation. Therefore, the accurate identification of TSSs is important for understanding the molecular regulation of transcription. Existing protocols for TSS identification are challenging and expensive, leaving high-quality data available for a small subset of organisms. This sparsity of data impairs study of TSS usage across tissues or in an evolutionary context. To address these shortcomings, we developed Smart-Seq2 Rolling Circle to Concatemeric Consensus (Smar2C2), which identifies and quantifies TSSs and transcription termination sites. Smar2C2 incorporates unique molecular identifiers that allowed for the identification of as many as 70 million sites, with no known upper limit. We have also generated TSS data sets from as little as 40 pg of total RNA, which was the smallest input tested. In this study, we used Smar2C2 to identify TSSs in Glycine max (soybean), Oryza sativa (rice), Sorghum bicolor (sorghum), Triticum aestivum (wheat) and Zea mays (maize) across multiple tissues. This wide panel of plant TSSs facilitated the identification of evolutionarily conserved features, such as novel patterns in the dinucleotides that compose the initiator element (Inr), that correlated with promoter expression levels across all species examined. We also discovered sequence variations in known promoter motifs that are positioned reliably close to the TSS, such as differences in the TATA box and in the Inr that may prove significant to our understanding and control of transcription initiation. Smar2C2 allows for the easy study of these critical sequences, providing a tool to facilitate discovery.
Keywords: cis-regulatory elements; promoter; rolling circle amplification; technical advance; template switching reverse transcriptase; transcription start site.
© 2022 The Authors. The Plant Journal published by Society for Experimental Biology and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that they have no conflicts of interest associated with this work.
Figures
Similar articles
-
Smar2C2: A Simple and Efficient Protocol for the Identification of Transcription Start Sites.Curr Protoc. 2023 Mar;3(3):e705. doi: 10.1002/cpz1.705. Curr Protoc. 2023. PMID: 36947693
-
Core Promoter Plasticity Between Maize Tissues and Genotypes Contrasts with Predominance of Sharp Transcription Initiation Sites.Plant Cell. 2015 Dec;27(12):3309-20. doi: 10.1105/tpc.15.00630. Epub 2015 Dec 1. Plant Cell. 2015. PMID: 26628745 Free PMC article.
-
Genome-wide computational prediction and analysis of core promoter elements across plant monocots and dicots.PLoS One. 2013 Oct 29;8(10):e79011. doi: 10.1371/journal.pone.0079011. eCollection 2013. PLoS One. 2013. PMID: 24205361 Free PMC article.
-
The RNA polymerase II core promoter - the gateway to transcription.Curr Opin Cell Biol. 2008 Jun;20(3):253-9. doi: 10.1016/j.ceb.2008.03.003. Epub 2008 Apr 22. Curr Opin Cell Biol. 2008. PMID: 18436437 Free PMC article. Review.
-
Global approaches for profiling transcription initiation.Cell Rep Methods. 2021 Sep 27;1(5):100081. doi: 10.1016/j.crmeth.2021.100081. Epub 2021 Sep 16. Cell Rep Methods. 2021. PMID: 34632443 Free PMC article. Review.
Cited by
-
Core promoterome of barley embryo.Comput Struct Biotechnol J. 2023 Dec 5;23:264-277. doi: 10.1016/j.csbj.2023.12.003. eCollection 2024 Dec. Comput Struct Biotechnol J. 2023. PMID: 38173877 Free PMC article.
-
An improved method for the highly specific detection of transcription start sites.Nucleic Acids Res. 2024 Jan 25;52(2):e7. doi: 10.1093/nar/gkad1116. Nucleic Acids Res. 2024. PMID: 37994784 Free PMC article.
-
Plant Promoters and Terminators for High-Precision Bioengineering.Biodes Res. 2023 Jul 7;5:0013. doi: 10.34133/bdr.0013. eCollection 2023. Biodes Res. 2023. PMID: 37849460 Free PMC article. Review.
-
Enhancers associated with unstable RNAs are rare in plants.Nat Plants. 2024 Aug;10(8):1246-1257. doi: 10.1038/s41477-024-01741-9. Epub 2024 Jul 30. Nat Plants. 2024. PMID: 39080503 Free PMC article.
-
Designing artificial synthetic promoters for accurate, smart, and versatile gene expression in plants.Plant Commun. 2023 Jul 10;4(4):100558. doi: 10.1016/j.xplc.2023.100558. Epub 2023 Feb 9. Plant Commun. 2023. PMID: 36760129 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
