

Infantile Sandhoff disease with ventricular septal defect: a case report
- PMID: 36002893
- PMCID: PMC9404584
- DOI: 10.1186/s13256-022-03550-0
Infantile Sandhoff disease with ventricular septal defect: a case report
Abstract
Background: Infantile Sandhoff disease is a rare inherited disorder that progressively destroys nerve cells in the brain and spinal cord, and is classified under lysosomal storage disorder. It is an autosomal recessive disorder of sphingolipid metabolism that results from deficiency of the lysosomal enzymes β-hexosaminidase A and B. The resultant accumulation of GM2 ganglioside within both gray matter nuclei and myelin sheaths of the white matter results in eventual severe neuronal dysfunction and neurodegeneration.
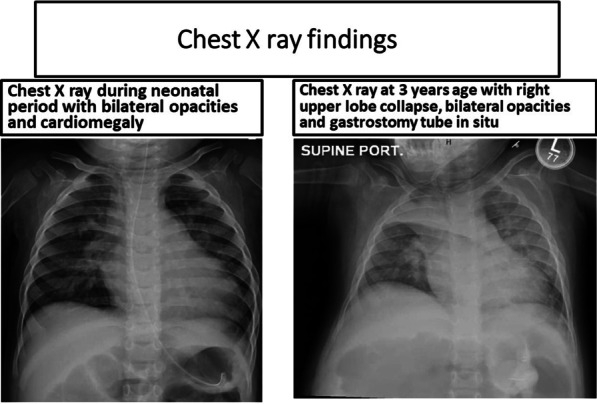
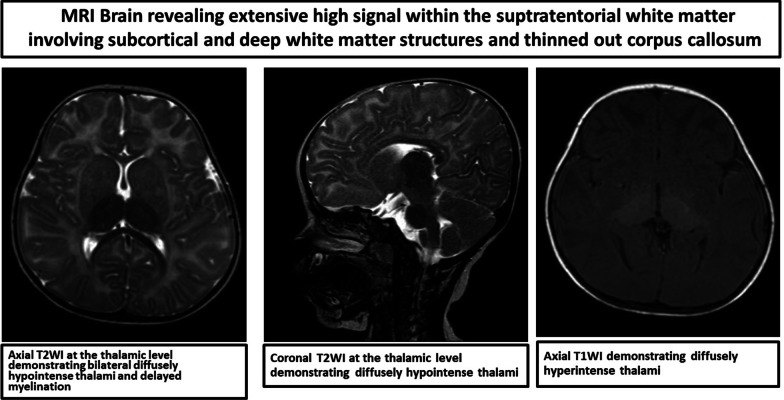
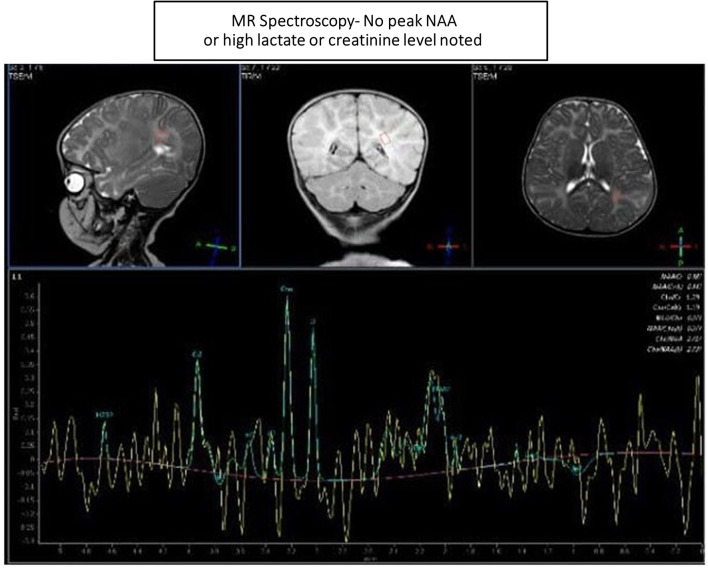
Case presentation: We evaluated a 3.5-year-old Comorian girl from the United Arab Emirates who presented with repeated chest infections with heart failure due to ventricular septal defect, neuroregression, recurrent seizures, and cherry-red spots over macula. She had macrocephaly, axial hypotonia, hyperacusis, and gastroesophageal reflux. Organomegaly was absent. Brain magnetic resonance imaging, metabolic tests, and genetic mutations confirmed the diagnosis. Despite multidisciplinary therapy, the girl succumbed to her illness.
Conclusion: Though early cardiac involvement can be seen with novel mutations, it is extremely rare to find association of ventricular septal defect in infantile Sandhoff disease. Neuroregression typically starts around 6 months of age. We report this case because of the unusual association of a congenital heart disease with underlying infantile Sandhoff disease and symptomatic heart failure in the first month of life with eventual fatal outcome.
Keywords: Cherry-red spots; Hexosaminidase; Hypotonia; Infantile Sandhoff disease; Neuroregression; Ventricular septal defect.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



Similar articles
-
Clinical presentation and outcome in infantile Sandhoff disease: a case series of 25 patients from Iranian neurometabolic bioregistry with five novel mutations.Orphanet J Rare Dis. 2018 Aug 3;13(1):130. doi: 10.1186/s13023-018-0876-5. Orphanet J Rare Dis. 2018. PMID: 30075786 Free PMC article.
-
Infantile onset Sandhoff disease: clinical manifestation and a novel common mutation in Thai patients.BMC Pediatr. 2021 Jan 7;21(1):22. doi: 10.1186/s12887-020-02481-3. BMC Pediatr. 2021. PMID: 33407268 Free PMC article.
-
Clinical and Molecular Characteristics of Two Chinese Children with Infantile Sandhoff Disease and Review of the Literature.J Mol Neurosci. 2020 Apr;70(4):481-487. doi: 10.1007/s12031-019-01409-6. Epub 2020 Jan 9. J Mol Neurosci. 2020. PMID: 31919734 Review.
-
[HEXB gene study and prenatal diagnosis for a family affected by infantile Sandhoff disease].Zhejiang Da Xue Xue Bao Yi Xue Ban. 2013 Jul;42(4):403-10. doi: 10.3785/j.issn.1008-9292.2013.04.006. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2013. PMID: 24022928 Chinese.
-
[Molecular pathogenesis and therapeutic approach of GM2 gangliosidosis].Yakugaku Zasshi. 2013;133(2):269-74. doi: 10.1248/yakushi.12-00199. Yakugaku Zasshi. 2013. PMID: 23370522 Review. Japanese.
Cited by
-
Infantile Neuroaxonal Dystrophy: Case Report and Review of Literature.Medicina (Kaunas). 2024 Aug 15;60(8):1322. doi: 10.3390/medicina60081322. Medicina (Kaunas). 2024. PMID: 39202603 Free PMC article. Review.
-
Molecular Basis of Müllerian Agenesis Causing Congenital Uterine Factor Infertility-A Systematic Review.Int J Mol Sci. 2023 Dec 21;25(1):120. doi: 10.3390/ijms25010120. Int J Mol Sci. 2023. PMID: 38203291 Free PMC article. Review.
-
Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review.Int J Mol Sci. 2023 May 11;24(10):8632. doi: 10.3390/ijms24108632. Int J Mol Sci. 2023. PMID: 37239976 Free PMC article. Review.
References
-
- Tavasoli AR, Parvaneh N, Ashrafi MR, Rezaei Z, Zschocke J, Rostami P. Clinical presentation and outcome in infantile Sandhoff disease: a case series of 25 patients from Iranian neurometabolic bioregistry with five novel mutations. Orphanet J Rare Dis. 2018;13(1):130. doi: 10.1186/s13023-018-0876-5. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
