

Regulated cell death (RCD) in cancer: key pathways and targeted therapies
- PMID: 35963853
- PMCID: PMC9376115
- DOI: 10.1038/s41392-022-01110-y
Regulated cell death (RCD) in cancer: key pathways and targeted therapies
Abstract
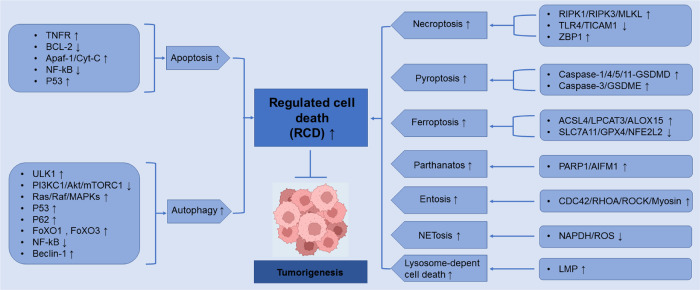
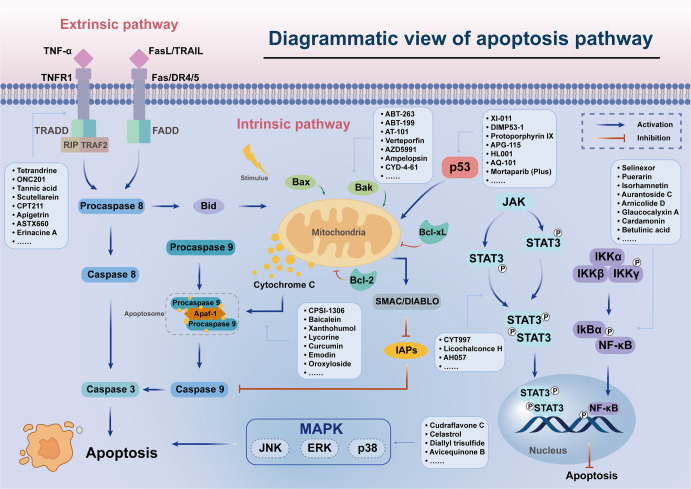
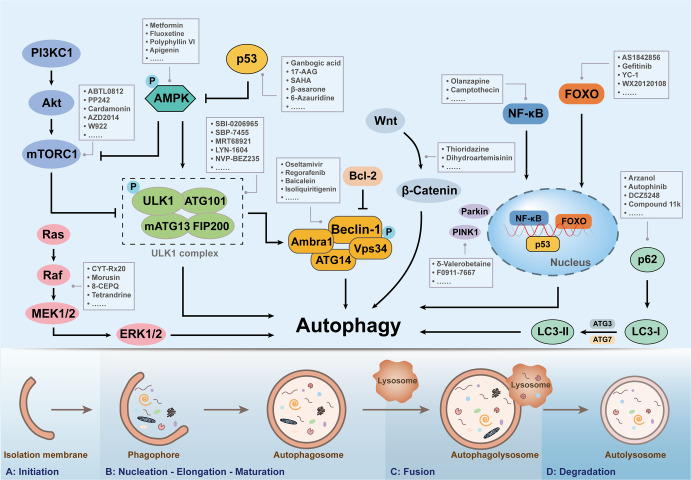
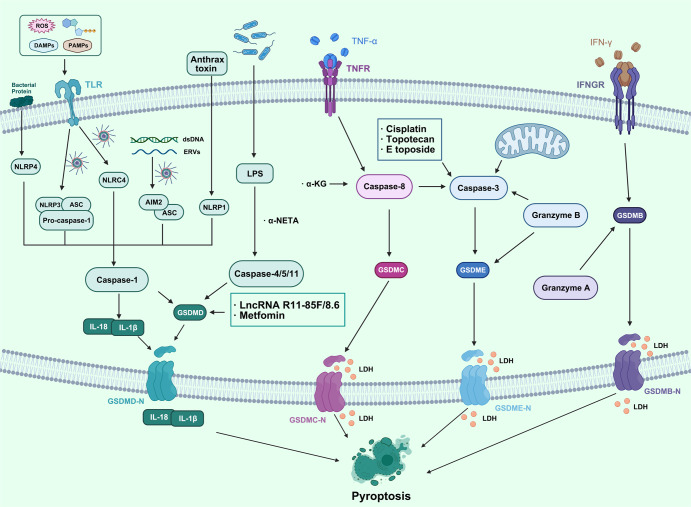
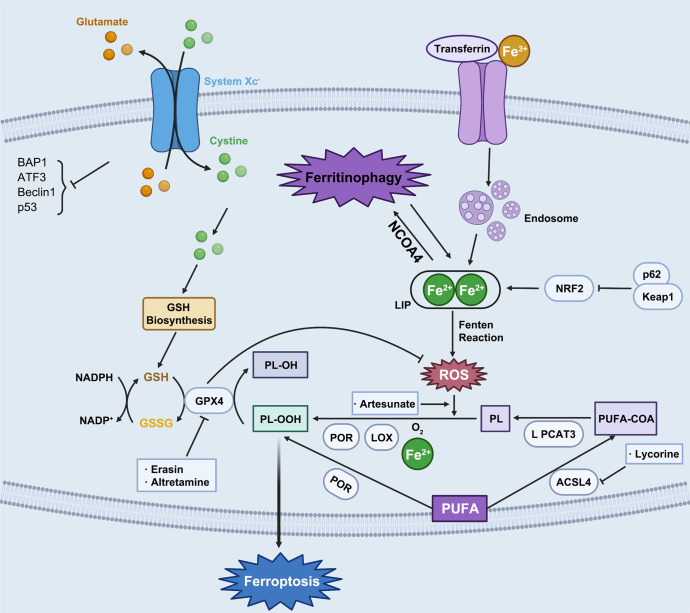
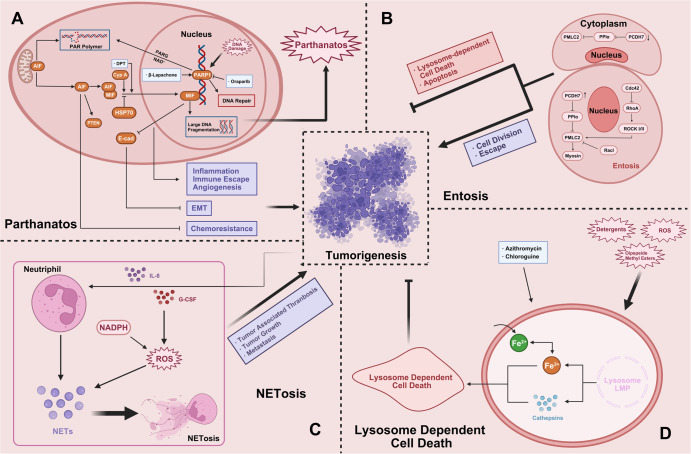
Regulated cell death (RCD), also well-known as programmed cell death (PCD), refers to the form of cell death that can be regulated by a variety of biomacromolecules, which is distinctive from accidental cell death (ACD). Accumulating evidence has revealed that RCD subroutines are the key features of tumorigenesis, which may ultimately lead to the establishment of different potential therapeutic strategies. Hitherto, targeting the subroutines of RCD with pharmacological small-molecule compounds has been emerging as a promising therapeutic avenue, which has rapidly progressed in many types of human cancers. Thus, in this review, we focus on summarizing not only the key apoptotic and autophagy-dependent cell death signaling pathways, but the crucial pathways of other RCD subroutines, including necroptosis, pyroptosis, ferroptosis, parthanatos, entosis, NETosis and lysosome-dependent cell death (LCD) in cancer. Moreover, we further discuss the current situation of several small-molecule compounds targeting the different RCD subroutines to improve cancer treatment, such as single-target, dual or multiple-target small-molecule compounds, drug combinations, and some new emerging therapeutic strategies that would together shed new light on future directions to attack cancer cell vulnerabilities with small-molecule drugs targeting RCD for therapeutic purposes.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: a revisited perspective from molecular mechanisms to targeted therapies.J Hematol Oncol. 2022 Apr 12;15(1):44. doi: 10.1186/s13045-022-01260-0. J Hematol Oncol. 2022. PMID: 35414025 Free PMC article. Review.
-
Therapeutic strategies of targeting non-apoptotic regulated cell death (RCD) with small-molecule compounds in cancer.Acta Pharm Sin B. 2024 Jul;14(7):2815-2853. doi: 10.1016/j.apsb.2024.04.020. Epub 2024 Apr 24. Acta Pharm Sin B. 2024. PMID: 39027232 Free PMC article. Review.
-
Emerging role of necroptosis, pyroptosis, and ferroptosis in breast cancer: New dawn for overcoming therapy resistance.Neoplasia. 2024 Sep;55:101017. doi: 10.1016/j.neo.2024.101017. Epub 2024 Jun 14. Neoplasia. 2024. PMID: 38878618 Free PMC article. Review.
-
Targeting regulated cell death with plant natural compounds for cancer therapy: A revisited review of apoptosis, autophagy-dependent cell death, and necroptosis.Phytother Res. 2023 Apr;37(4):1488-1525. doi: 10.1002/ptr.7738. Epub 2023 Jan 30. Phytother Res. 2023. PMID: 36717200 Review.
-
Targeting regulated cell death (RCD) with small-molecule compounds in cancer therapy: A revisited review of apoptosis, autophagy-dependent cell death and necroptosis.Drug Discov Today. 2022 Feb;27(2):612-625. doi: 10.1016/j.drudis.2021.10.011. Epub 2021 Oct 27. Drug Discov Today. 2022. PMID: 34718209 Review.
Cited by
-
Disulfidptosis: disulfide stress-induced novel cell death pathway.MedComm (2020). 2024 Jun 29;5(7):e579. doi: 10.1002/mco2.579. eCollection 2024 Jul. MedComm (2020). 2024. PMID: 38948113 Free PMC article.
-
Targeting endocytosis to sensitize cancer cells to programmed cell death.Biochem Soc Trans. 2024 Aug 28;52(4):1703-1713. doi: 10.1042/BST20231332. Biochem Soc Trans. 2024. PMID: 39092762 Free PMC article. Review.
-
Comprehensive characterization of β-alanine metabolism-related genes in HCC identified a novel prognostic signature related to clinical outcomes.Aging (Albany NY). 2024 Apr 16;16(8):7073-7100. doi: 10.18632/aging.205744. Epub 2024 Apr 16. Aging (Albany NY). 2024. PMID: 38637116 Free PMC article.
-
Machine learning-based construction of immunogenic cell death-related score for improving prognosis and response to immunotherapy in melanoma.Aging (Albany NY). 2023 Apr 6;15(7):2667-2688. doi: 10.18632/aging.204636. Epub 2023 Apr 6. Aging (Albany NY). 2023. PMID: 37036471 Free PMC article.
-
Constructing a Prognostic Model of Uterine Corpus Endometrial Carcinoma and Predicting Drug-Sensitivity Responses Using Programmed Cell Death-Related Pathways.J Cancer. 2024 Mar 31;15(10):2948-2959. doi: 10.7150/jca.92201. eCollection 2024. J Cancer. 2024. PMID: 38706893 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
