

Conformational buffering underlies functional selection in intrinsically disordered protein regions
- PMID: 35948766
- PMCID: PMC10262780
- DOI: 10.1038/s41594-022-00811-w
Conformational buffering underlies functional selection in intrinsically disordered protein regions
Abstract
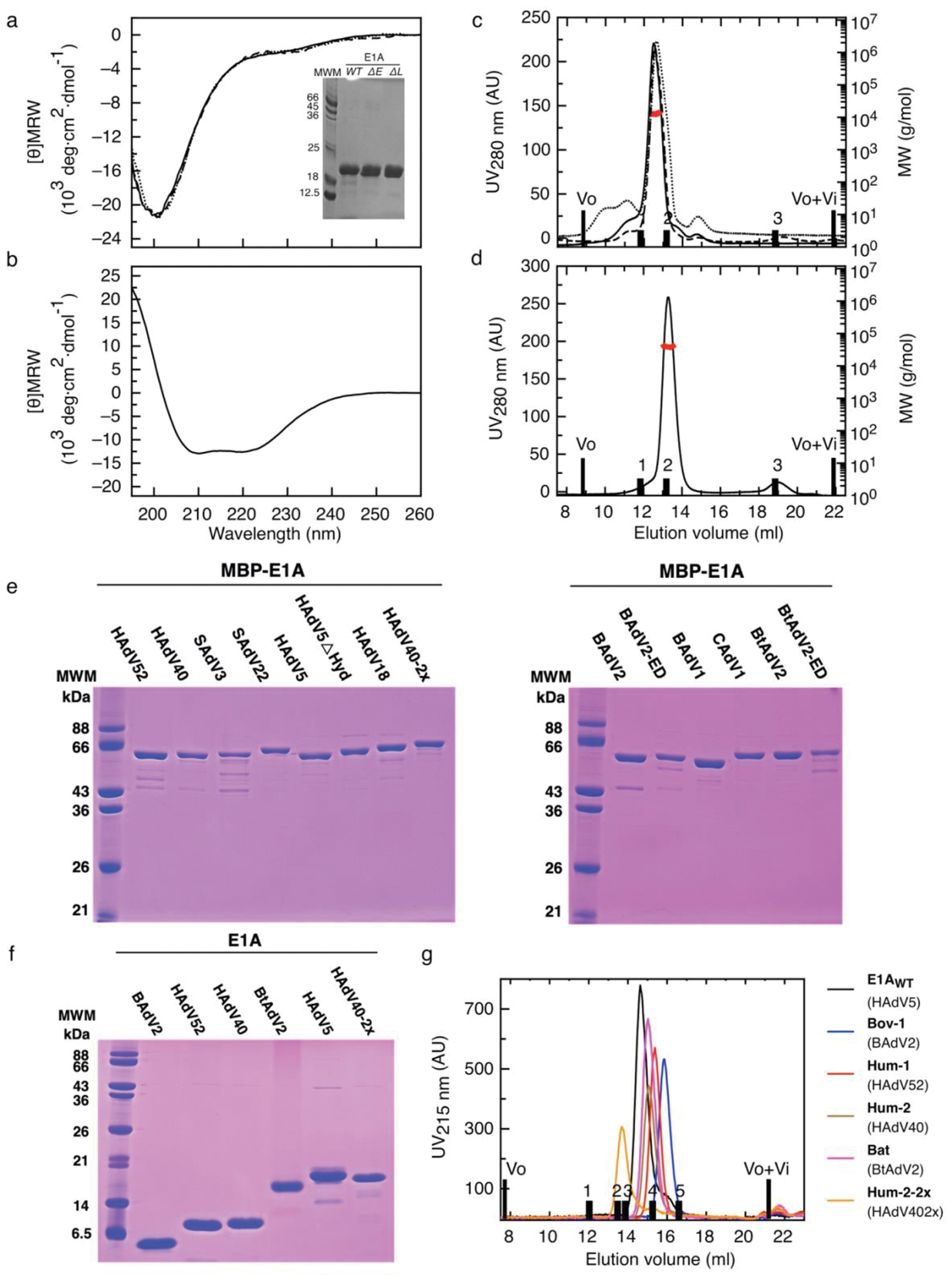
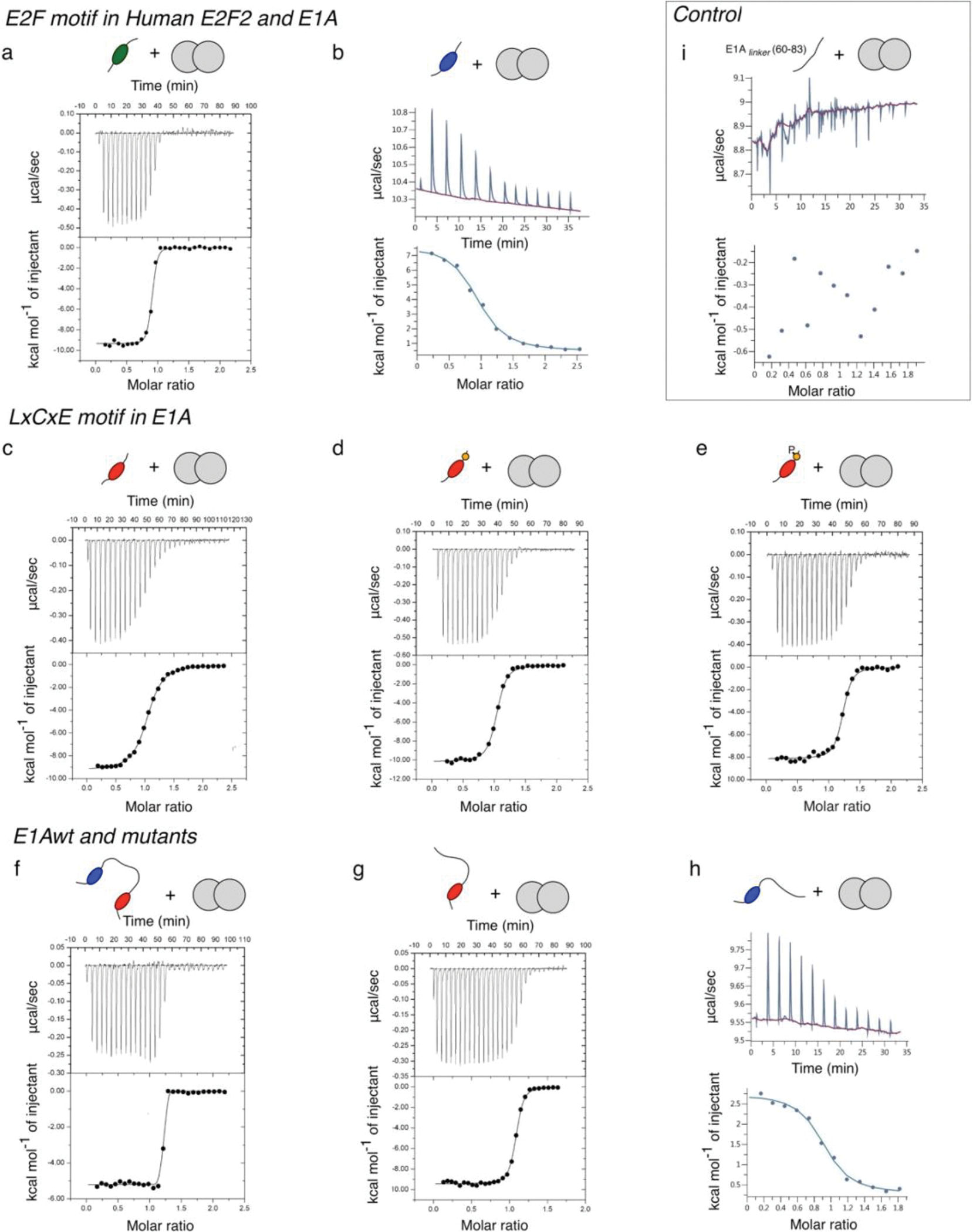
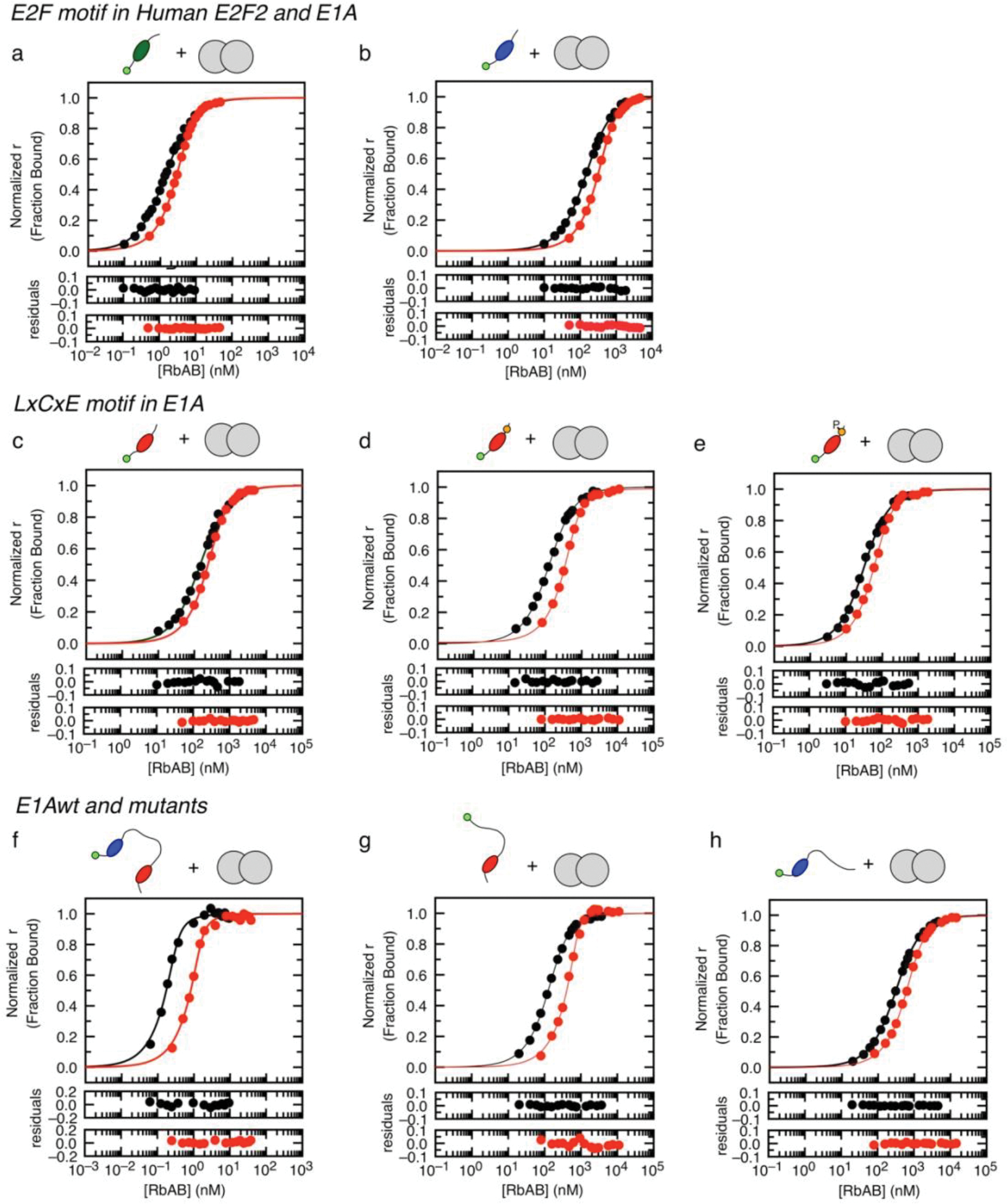
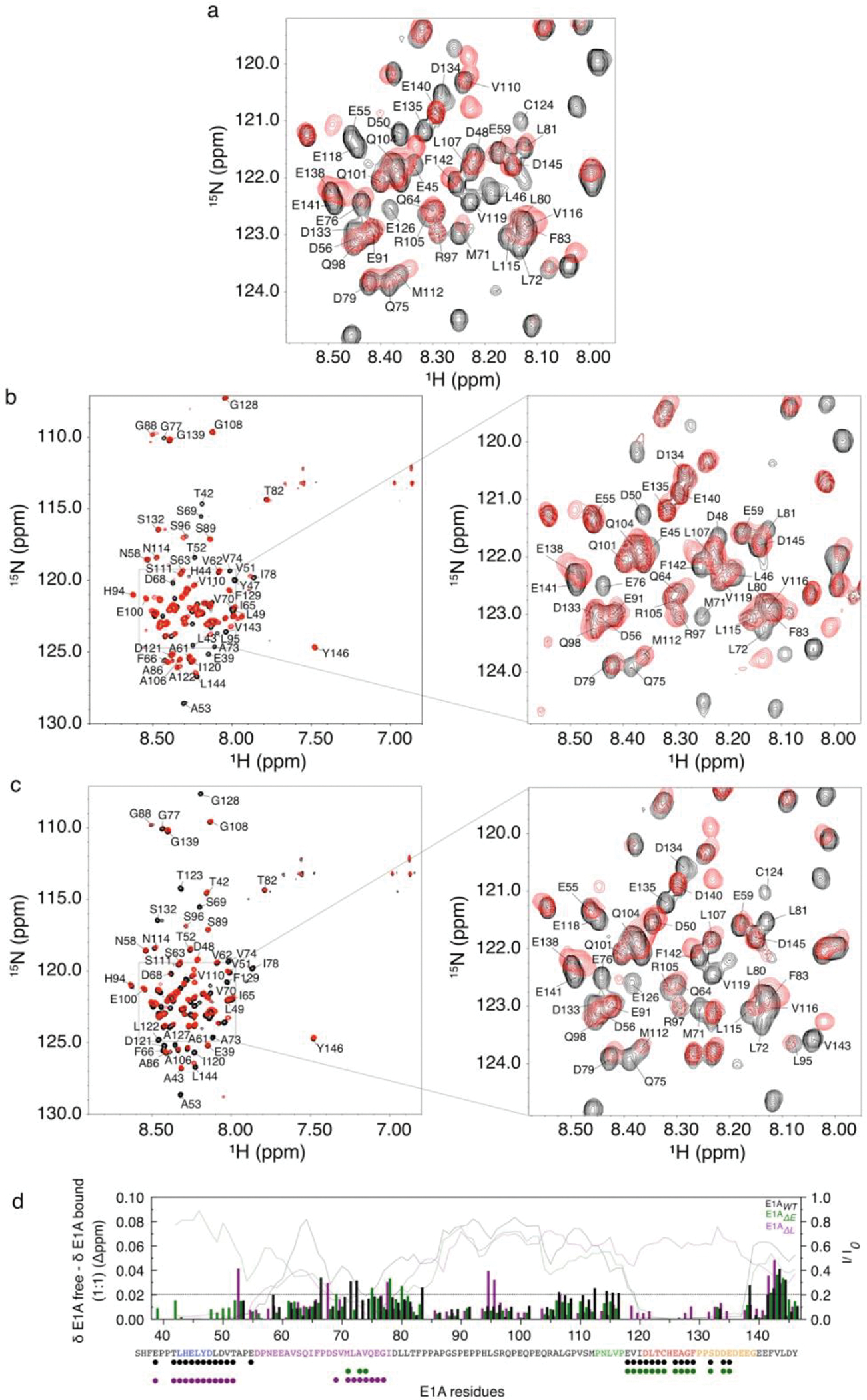
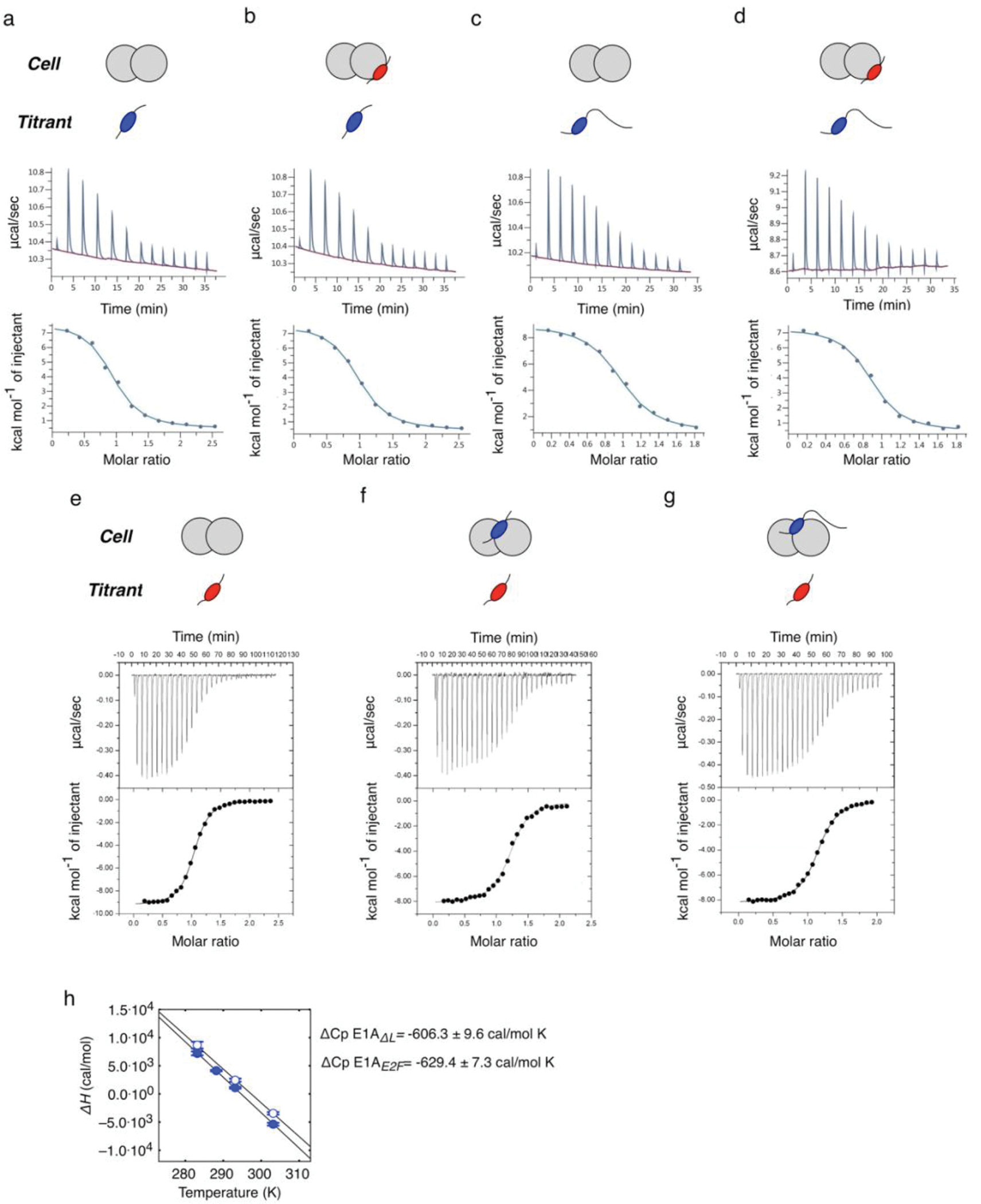
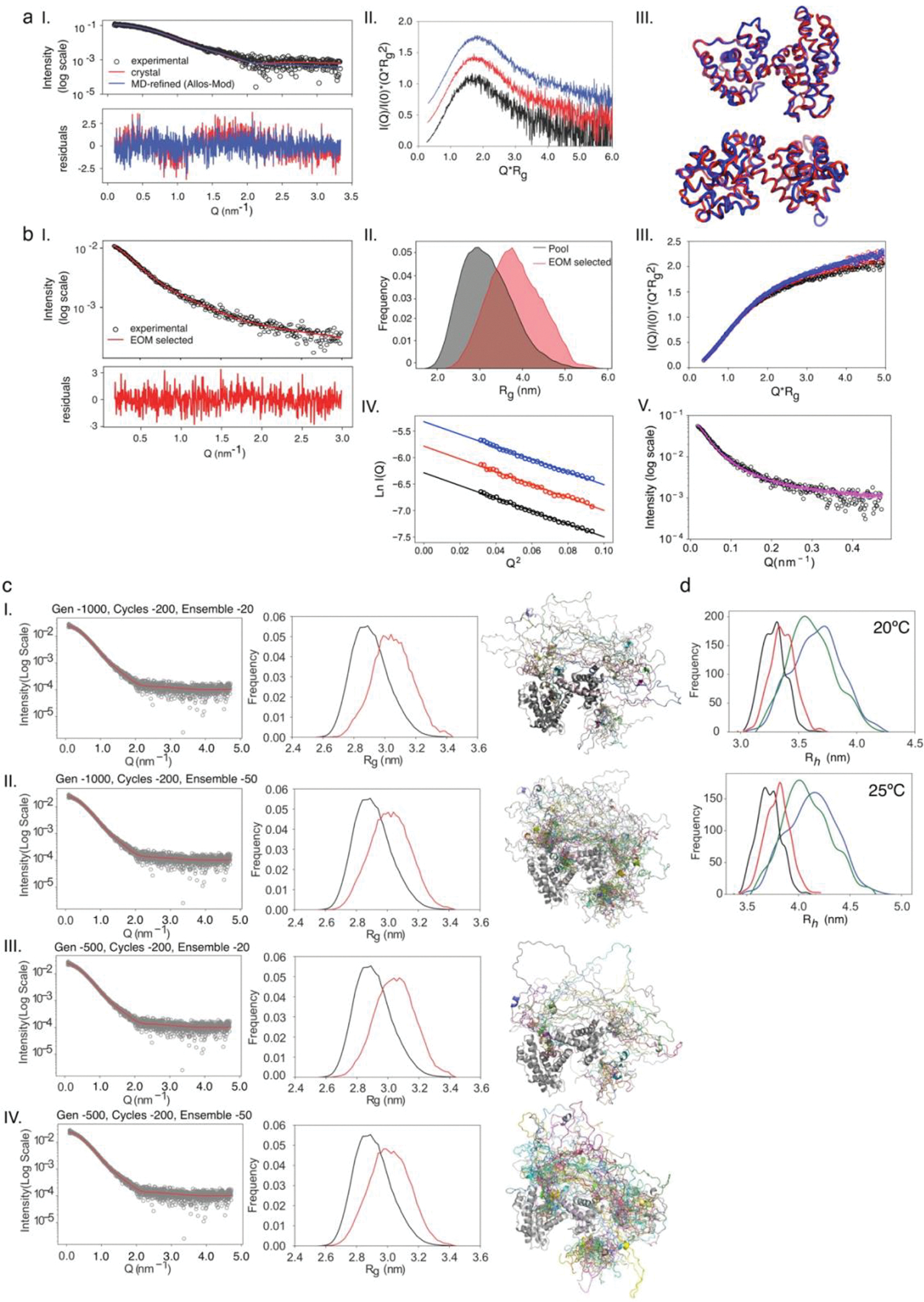
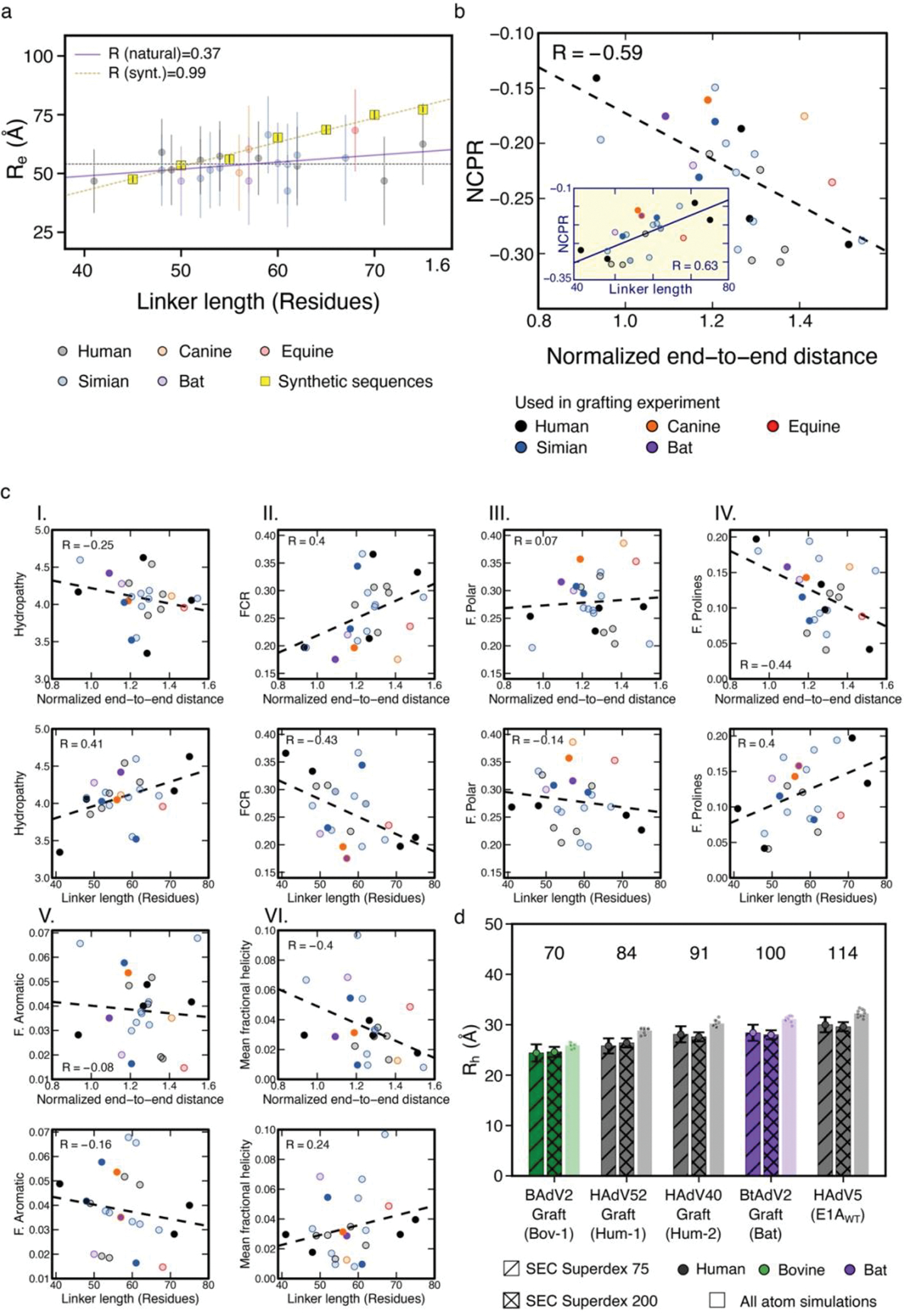
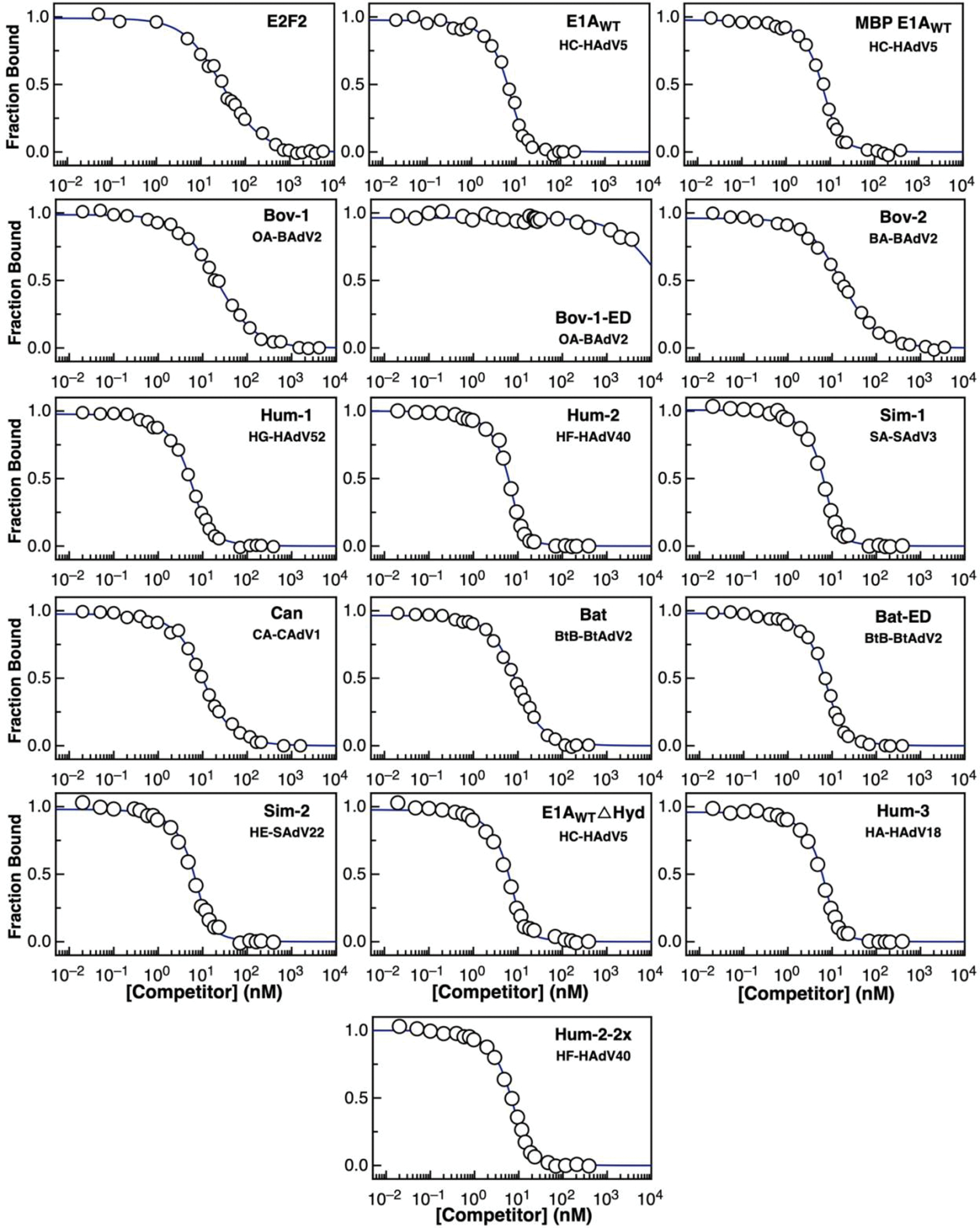
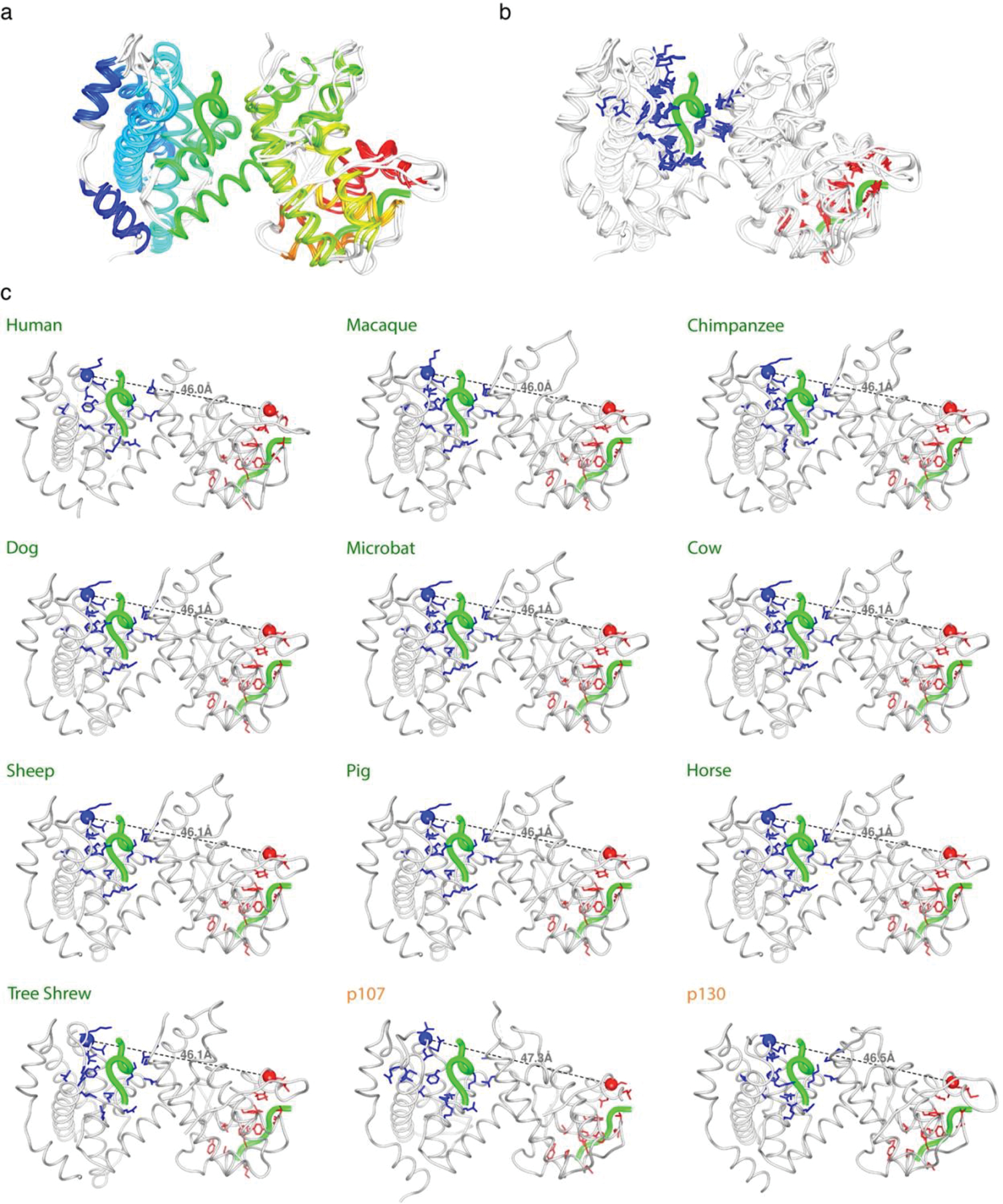
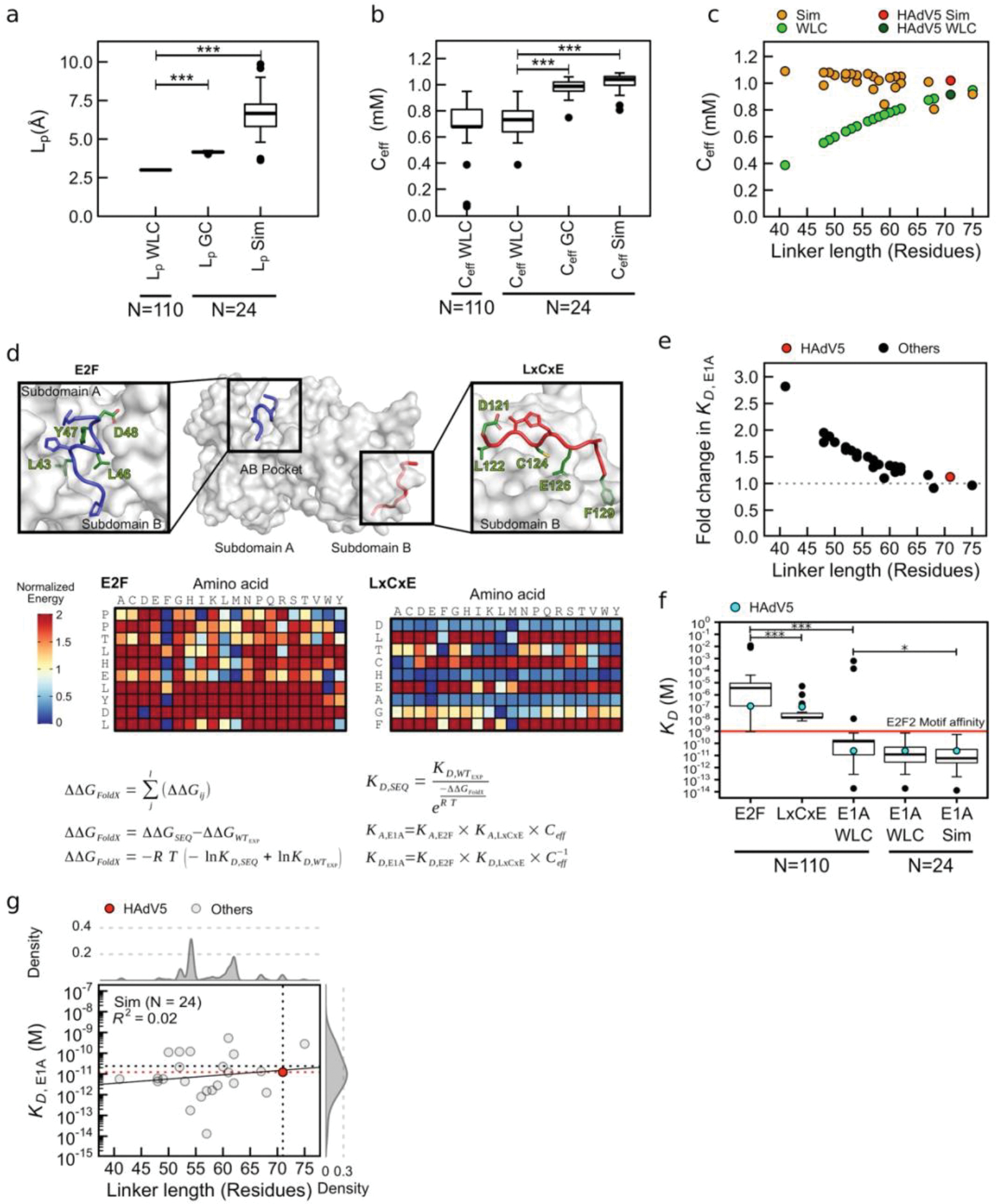
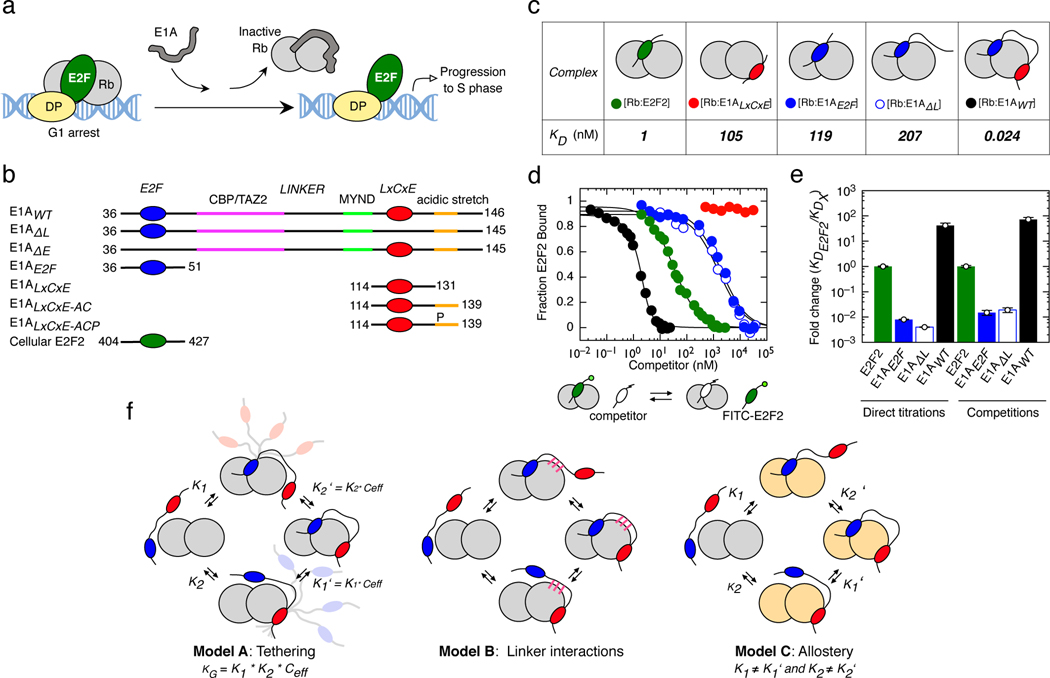
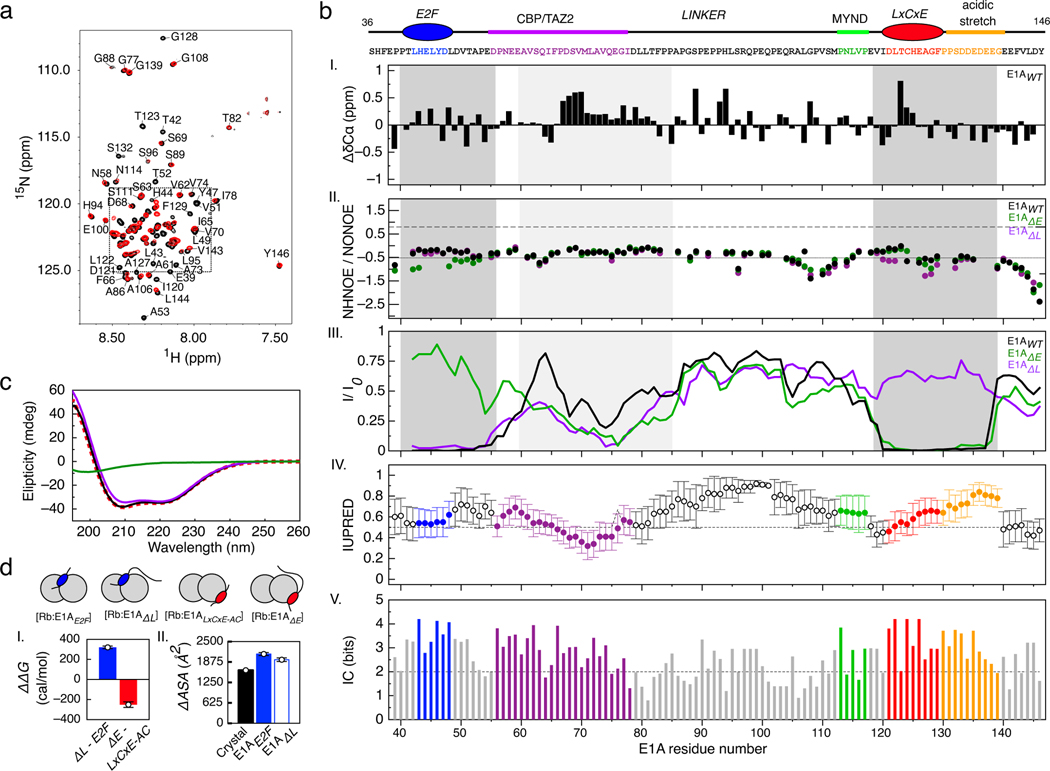
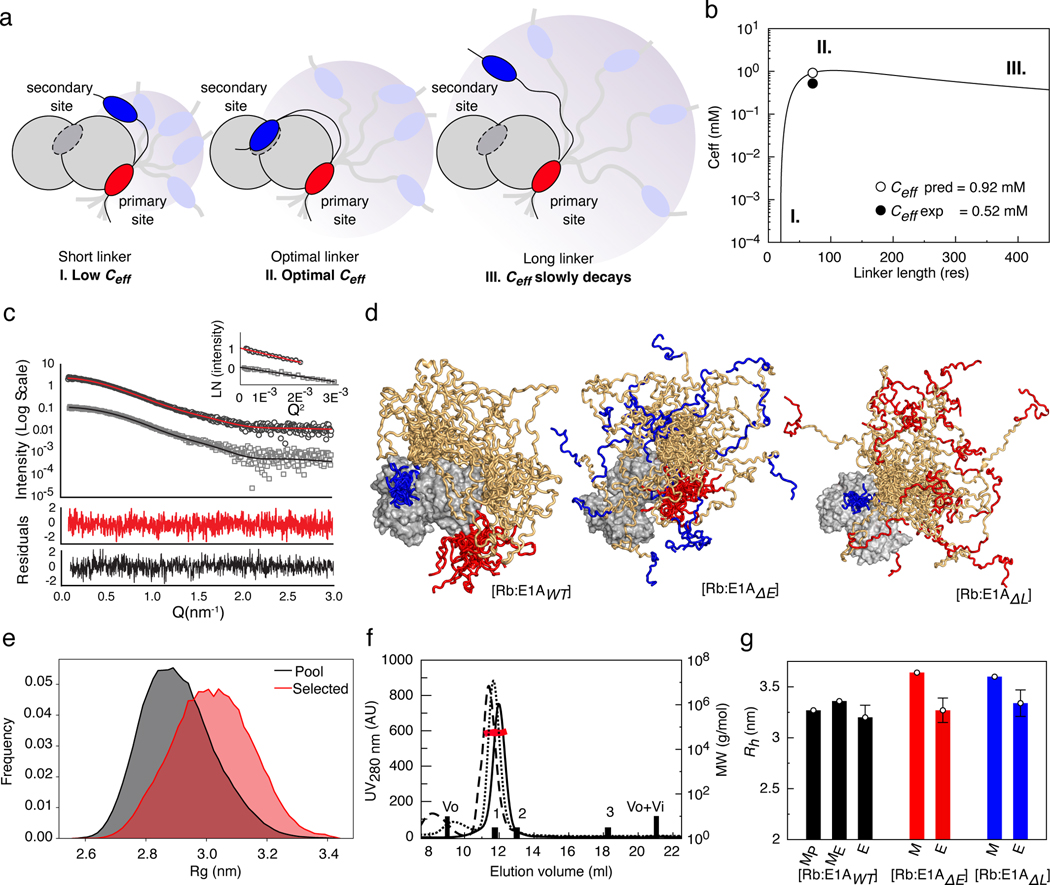
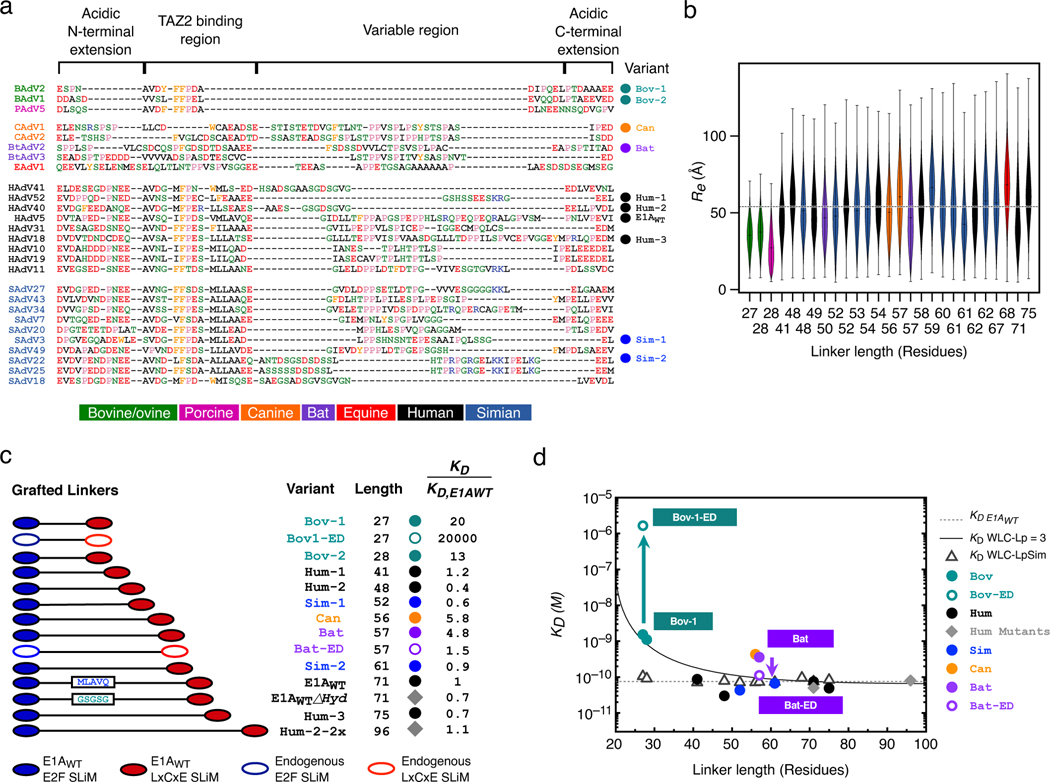
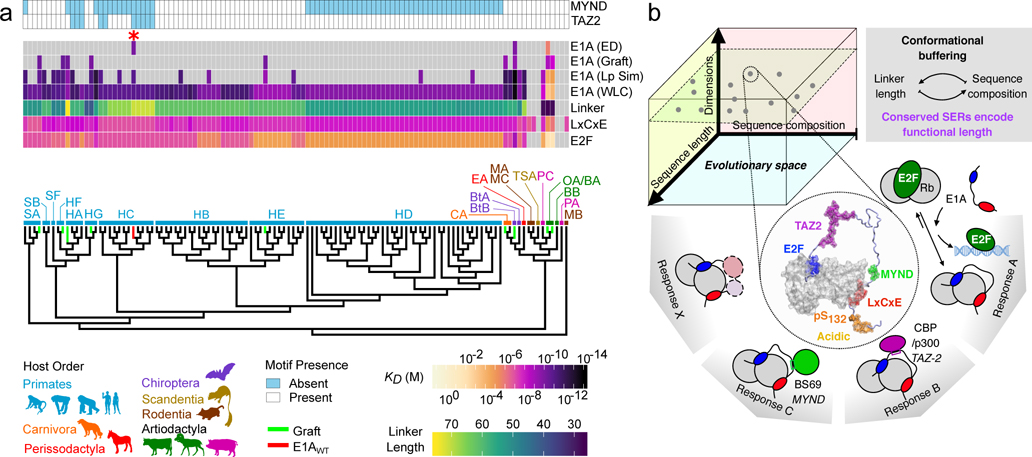
Many disordered proteins conserve essential functions in the face of extensive sequence variation, making it challenging to identify the mechanisms responsible for functional selection. Here we identify the molecular mechanism of functional selection for the disordered adenovirus early gene 1A (E1A) protein. E1A competes with host factors to bind the retinoblastoma (Rb) protein, subverting cell cycle regulation. We show that two binding motifs tethered by a hypervariable disordered linker drive picomolar affinity Rb binding and host factor displacement. Compensatory changes in amino acid sequence composition and sequence length lead to conservation of optimal tethering across a large family of E1A linkers. We refer to this compensatory mechanism as conformational buffering. We also detect coevolution of the motifs and linker, which can preserve or eliminate the tethering mechanism. Conformational buffering and motif-linker coevolution explain robust functional encoding within hypervariable disordered linkers and could underlie functional selection of many disordered protein regions.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
A.S.H. is a scientific consultant with Dewpoint Therapeutics Inc. and R.V.P. is a member of the scientific advisory board of Dewpoint Therapeutics Inc. This work has not been influenced by the affiliation with Dewpoint. The rest of the authors have no competing interests.
Figures
Similar articles
-
Modulation of allostery by protein intrinsic disorder.Nature. 2013 Jun 20;498(7454):390-4. doi: 10.1038/nature12294. Nature. 2013. PMID: 23783631 Free PMC article.
-
Interplay between sequence, structure and linear motifs in the adenovirus E1A hub protein.Virology. 2018 Dec;525:117-131. doi: 10.1016/j.virol.2018.08.012. Epub 2018 Sep 25. Virology. 2018. PMID: 30265888
-
Adenoviral E1A Exploits Flexibility and Disorder to Target Cellular Proteins.Biomolecules. 2020 Nov 11;10(11):1541. doi: 10.3390/biom10111541. Biomolecules. 2020. PMID: 33187345 Free PMC article.
-
Adenovirus-5 E1A: paradox and paradigm.Nat Rev Mol Cell Biol. 2002 Jun;3(6):441-52. doi: 10.1038/nrm827. Nat Rev Mol Cell Biol. 2002. PMID: 12042766 Review.
-
How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40.Virology. 2009 Feb 20;384(2):274-84. doi: 10.1016/j.virol.2008.12.010. Epub 2009 Jan 17. Virology. 2009. PMID: 19150725 Review.
Cited by
-
The molecular basis for cellular function of intrinsically disordered protein regions.Nat Rev Mol Cell Biol. 2024 Mar;25(3):187-211. doi: 10.1038/s41580-023-00673-0. Epub 2023 Nov 13. Nat Rev Mol Cell Biol. 2024. PMID: 37957331 Free PMC article. Review.
-
Systematic identification of conditionally folded intrinsically disordered regions by AlphaFold2.Proc Natl Acad Sci U S A. 2023 Oct 31;120(44):e2304302120. doi: 10.1073/pnas.2304302120. Epub 2023 Oct 25. Proc Natl Acad Sci U S A. 2023. PMID: 37878721 Free PMC article.
-
KMT5C leverages disorder to optimize cooperation with HP1 for heterochromatin retention.EMBO Rep. 2025 Jan;26(1):153-174. doi: 10.1038/s44319-024-00320-5. Epub 2024 Nov 19. EMBO Rep. 2025. PMID: 39562713 Free PMC article.
-
Sequence-ensemble-function relationships for disordered proteins in live cells.bioRxiv [Preprint]. 2023 Nov 13:2023.10.29.564547. doi: 10.1101/2023.10.29.564547. bioRxiv. 2023. PMID: 37961106 Free PMC article. Preprint.
-
The MDMX Acidic Domain Uses Allovalency to Bind Both p53 and MDMX.J Mol Biol. 2022 Nov 30;434(22):167844. doi: 10.1016/j.jmb.2022.167844. Epub 2022 Sep 29. J Mol Biol. 2022. PMID: 36181774 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
