

G9a dictates neuronal vulnerability to inflammatory stress via transcriptional control of ferroptosis
- PMID: 35930635
- PMCID: PMC9355351
- DOI: 10.1126/sciadv.abm5500
G9a dictates neuronal vulnerability to inflammatory stress via transcriptional control of ferroptosis
Abstract
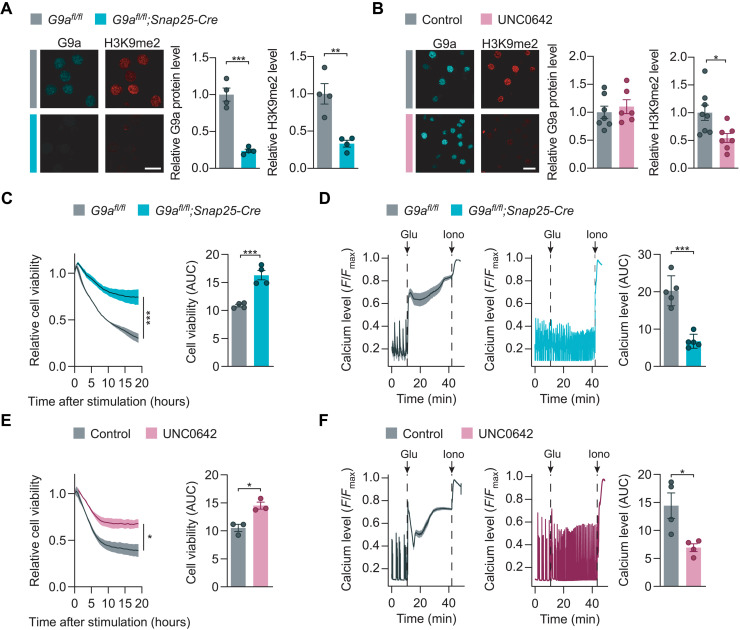
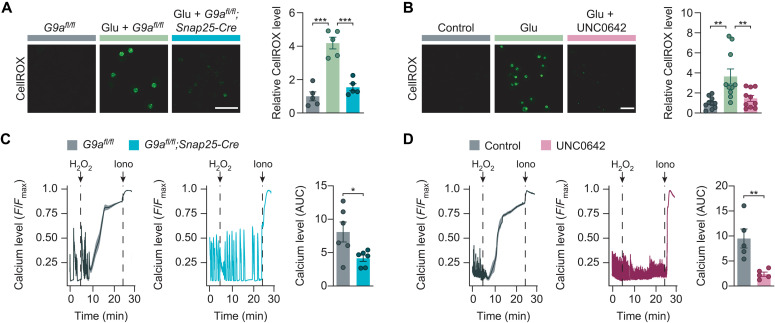
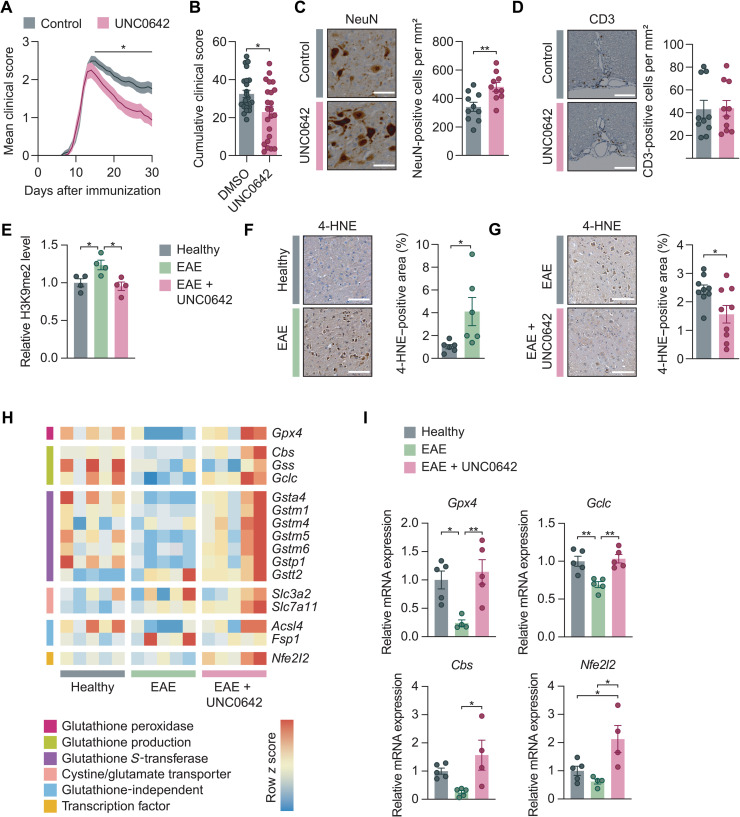
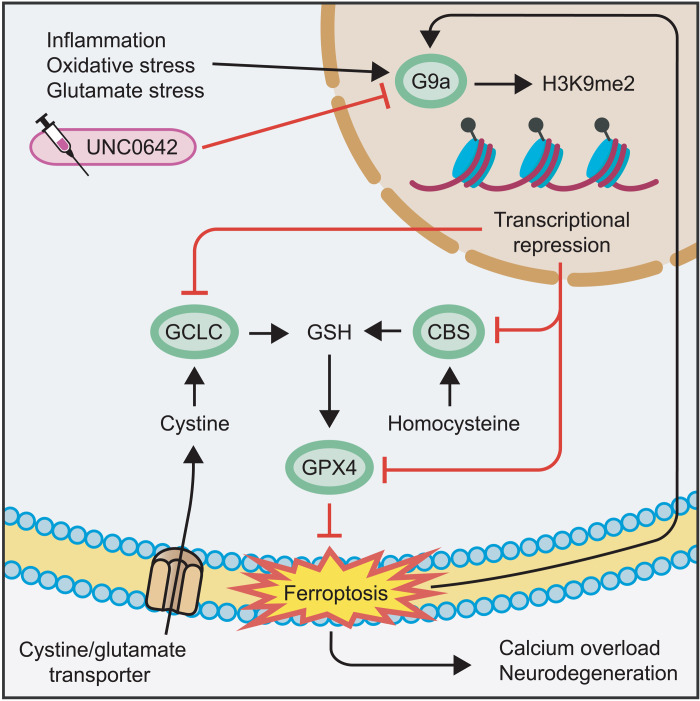
Neuroinflammation leads to neuronal stress responses that contribute to neuronal dysfunction and loss. However, treatments that stabilize neurons and prevent their destruction are still lacking. Here, we identify the histone methyltransferase G9a as a druggable epigenetic regulator of neuronal vulnerability to inflammation. In murine experimental autoimmune encephalomyelitis (EAE) and human multiple sclerosis (MS), we found that the G9a-catalyzed repressive epigenetic mark H3K9me2 was robustly induced by neuroinflammation. G9a activity repressed anti-ferroptotic genes, diminished intracellular glutathione levels, and triggered the iron-dependent programmed cell death pathway ferroptosis. Conversely, pharmacological treatment of EAE mice with a G9a inhibitor restored anti-ferroptotic gene expression, reduced inflammation-induced neuronal loss, and improved clinical outcome. Similarly, neuronal anti-ferroptotic gene expression was reduced in MS brain tissue and was boosted by G9a inhibition in human neuronal cultures. This study identifies G9a as a critical transcriptional enhancer of neuronal ferroptosis and potential therapeutic target to counteract inflammation-induced neurodegeneration.
Figures



Similar articles
-
Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice.Neuroscience. 2014 Jan 31;258:422-32. doi: 10.1016/j.neuroscience.2013.11.043. Epub 2013 Dec 1. Neuroscience. 2014. PMID: 24300108 Free PMC article.
-
Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis.J Neurochem. 2019 Feb;148(3):426-439. doi: 10.1111/jnc.14604. Epub 2018 Dec 3. J Neurochem. 2019. PMID: 30289974 Free PMC article.
-
Recruitment of histone methyltransferase G9a mediates transcriptional repression of Fgf21 gene by E4BP4 protein.J Biol Chem. 2013 Feb 22;288(8):5417-25. doi: 10.1074/jbc.M112.433482. Epub 2013 Jan 2. J Biol Chem. 2013. PMID: 23283977 Free PMC article.
-
The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function.Front Immunol. 2017 Apr 11;8:429. doi: 10.3389/fimmu.2017.00429. eCollection 2017. Front Immunol. 2017. PMID: 28443098 Free PMC article. Review.
-
H3K9 methyltransferase G9a and the related molecule GLP.Genes Dev. 2011 Apr 15;25(8):781-8. doi: 10.1101/gad.2027411. Genes Dev. 2011. PMID: 21498567 Free PMC article. Review.
Cited by
-
Advances in research on immunocyte iron metabolism, ferroptosis, and their regulatory roles in autoimmune and autoinflammatory diseases.Cell Death Dis. 2024 Jul 4;15(7):481. doi: 10.1038/s41419-024-06807-2. Cell Death Dis. 2024. PMID: 38965216 Free PMC article. Review.
-
Effects of DNA, RNA, and Protein Methylation on the Regulation of Ferroptosis.Int J Biol Sci. 2023 Jul 9;19(11):3558-3575. doi: 10.7150/ijbs.85454. eCollection 2023. Int J Biol Sci. 2023. PMID: 37497000 Free PMC article. Review.
-
Emerging role of ferroptosis in metabolic dysfunction-associated steatotic liver disease: revisiting hepatic lipid peroxidation.EBioMedicine. 2024 Apr;102:105088. doi: 10.1016/j.ebiom.2024.105088. Epub 2024 Mar 26. EBioMedicine. 2024. PMID: 38537604 Free PMC article. Review.
-
Spatial multi-omics: deciphering technological landscape of integration of multi-omics and its applications.J Hematol Oncol. 2024 Aug 24;17(1):72. doi: 10.1186/s13045-024-01596-9. J Hematol Oncol. 2024. PMID: 39182134 Free PMC article. Review.
-
Loss of renal tubular G9a benefits acute kidney injury by lowering focal lipid accumulation via CES1.EMBO Rep. 2023 Jun 5;24(6):e56128. doi: 10.15252/embr.202256128. Epub 2023 Apr 12. EMBO Rep. 2023. PMID: 37042626 Free PMC article.
References
-
- Friese M. A., Schattling B., Fugger L., Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 10, 225–238 (2014). - PubMed
-
- Dendrou C. A., Fugger L., Friese M. A., Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558 (2015). - PubMed
-
- Luchetti S., Fransen N. L., van Eden C. G., Ramaglia V., Mason M., Huitinga I., Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: A retrospective autopsy cohort analysis. Acta Neuropathol. 135, 511–528 (2018). - PMC - PubMed
-
- Ransohoff R. M., How neuroinflammation contributes to neurodegeneration. Science 353, 777–783 (2016). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
