

Loss of tau expression attenuates neurodegeneration associated with α-synucleinopathy
- PMID: 35773715
- PMCID: PMC9248195
- DOI: 10.1186/s40035-022-00309-x
Loss of tau expression attenuates neurodegeneration associated with α-synucleinopathy
Abstract
Background: Neuronal dysfunction and degeneration linked to α-synuclein (αS) pathology is thought to be responsible for the progressive nature of Parkinson's disease and related dementia with Lewy bodies. Studies have indicated bidirectional pathological relationships between αS pathology and tau abnormalities. We recently showed that A53T mutant human αS (HuαS) can cause post-synaptic and cognitive deficits that require microtubule-associated protein tau expression. However, the role of tau in the development of αS pathology and subsequent neuronal dysfunction has been controversial. Herein, we set out to determine the role of tau in the onset and progression of αS pathology (α-synucleinopathy) using a transgenic mouse model of α-synucleinopathy lacking mouse tau expression.
Methods: Transgenic mice expressing A53T mutant HuαS (TgA53T) were crossed with mTau-/- mice to generate TgA53T/mTau-/-. To achieve more uniform induction of α-synucleinopathy in mice, we used intramuscular injections of αS preformed fibrils (PFF) in non-transgenic (nTg), TgA53T, TgA53T/mTau-/-, and mTau-/- mice. Motor behavior was analyzed at 70 days post inoculation (dpi) of PFF and tissues for biochemical and neuropathological analysis were collected at 40 dpi, 70 dpi, and end stage.
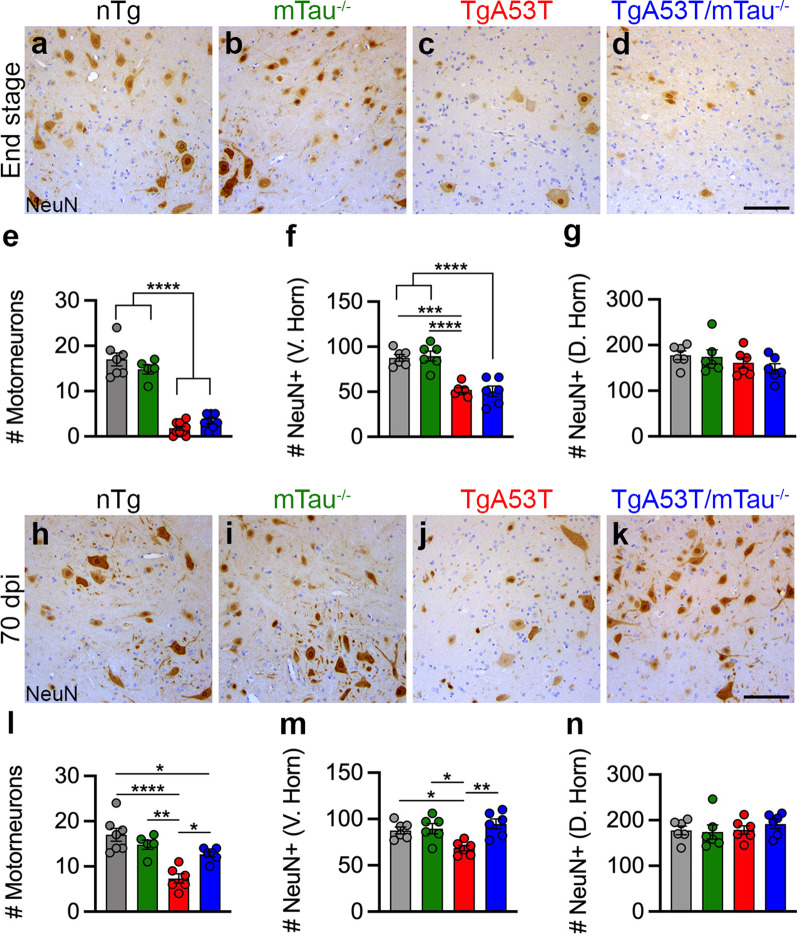
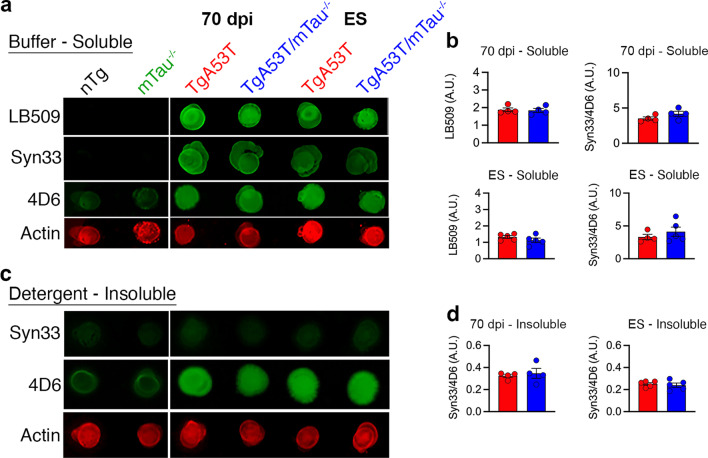
Results: Loss of tau expression significantly delayed the onset of motor deficits in the TgA53T model and the progression of α-synucleinopathy disease, as evidenced by a significant reduction in histopathological and behavioral markers of neurodegeneration and disease, and a significant improvement in survival. In vitro application of PFF to primary mouse hippocampal neurons demonstrated no changes in PFF uptake and processing or pS129 αS aggregation as a function of tau expression. However, PFF-induced neurotoxicity, including morphological deficits in nTg neurons, was prevented with tau removal.
Conclusions: Collectively, our data suggest that tau is likely acting downstream of αS pathology to affect neuronal homeostasis and survival. This work further supports the investigation of tau in α-synucleinopathies to identify novel disease-modifying therapeutic strategies.
Keywords: Lewy body disease; Neurodegeneration; Parkinson’s disease; Tau; α-Synuclein.
© 2022. The Author(s).
Conflict of interest statement
All experimental protocols involving mice were in strict adherence to the NIH Animal Care and Guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota. All applicable ethical standards required by University of Minnesota IACUC was followed. All authors declare no conflict of interest.
The authors declare no conflict of interest.
Figures






Similar articles
-
Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of α-synucleinopathy.Acta Neuropathol. 2019 Oct;138(4):551-574. doi: 10.1007/s00401-019-02032-w. Epub 2019 Jun 6. Acta Neuropathol. 2019. PMID: 31168644 Free PMC article.
-
α-Synucleinopathy associated c-Abl activation causes p53-dependent autophagy impairment.Mol Neurodegener. 2020 Apr 16;15(1):27. doi: 10.1186/s13024-020-00364-w. Mol Neurodegener. 2020. PMID: 32299471 Free PMC article.
-
Prominent microglial inclusions in transgenic mouse models of α-synucleinopathy that are distinct from neuronal lesions.Acta Neuropathol Commun. 2020 Aug 12;8(1):133. doi: 10.1186/s40478-020-00993-8. Acta Neuropathol Commun. 2020. PMID: 32787922 Free PMC article.
-
Vesicle trafficking and lipid metabolism in synucleinopathy.Acta Neuropathol. 2021 Apr;141(4):491-510. doi: 10.1007/s00401-020-02177-z. Epub 2020 Jun 30. Acta Neuropathol. 2021. PMID: 32607605 Free PMC article. Review.
-
α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies.Neurosci Lett. 2019 Sep 14;709:134316. doi: 10.1016/j.neulet.2019.134316. Epub 2019 Jun 3. Neurosci Lett. 2019. PMID: 31170426 Free PMC article. Review.
Cited by
-
Advances in understanding the function of alpha-synuclein: implications for Parkinson's disease.Brain. 2023 Sep 1;146(9):3587-3597. doi: 10.1093/brain/awad150. Brain. 2023. PMID: 37183455 Free PMC article. Review.
-
The role of exosomes in the diagnosis of Parkinson's disease.Heliyon. 2023 Oct 16;9(10):e20595. doi: 10.1016/j.heliyon.2023.e20595. eCollection 2023 Oct. Heliyon. 2023. PMID: 37928387 Free PMC article. Review.
-
Investigating the Pathogenic Interplay of Alpha-Synuclein, Tau, and Amyloid Beta in Lewy Body Dementia: Insights from Viral-Mediated Overexpression in Transgenic Mouse Models.Biomedicines. 2023 Oct 22;11(10):2863. doi: 10.3390/biomedicines11102863. Biomedicines. 2023. PMID: 37893236 Free PMC article.
-
Internalized α-synuclein fibrils become truncated and resist degradation in neurons while glial cells rapidly degrade α-synuclein fibrils.bioRxiv [Preprint]. 2024 Jun 8:2024.06.05.597615. doi: 10.1101/2024.06.05.597615. bioRxiv. 2024. PMID: 38895363 Free PMC article. Preprint.
-
The potential of exosomal biomarkers: Revolutionizing Parkinson's disease: How do they influence pathogenesis, diagnosis, and therapeutic strategies?AIMS Neurosci. 2024 Sep 23;11(3):374-397. doi: 10.3934/Neuroscience.2024023. eCollection 2024. AIMS Neurosci. 2024. PMID: 39431275 Free PMC article. Review.
References
-
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, et al. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
