

The insulin receptor family in the heart: new light on old insights
- PMID: 35766350
- PMCID: PMC9297685
- DOI: 10.1042/BSR20221212
The insulin receptor family in the heart: new light on old insights
Abstract
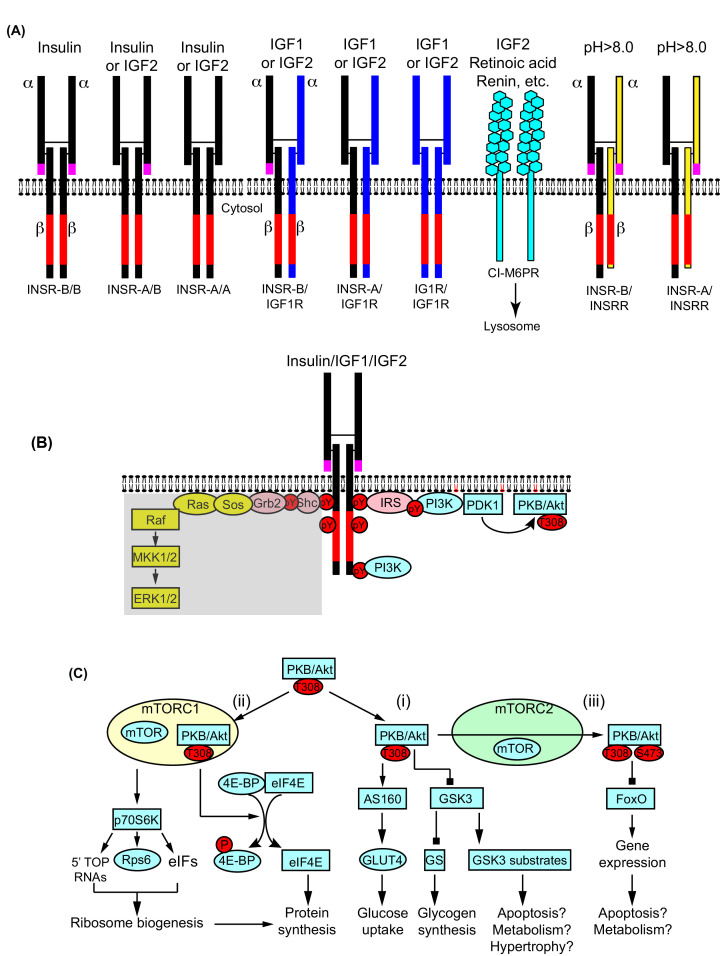
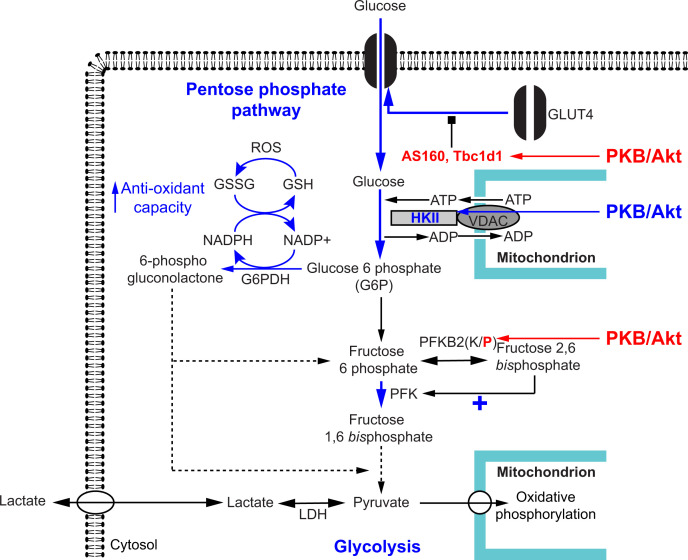
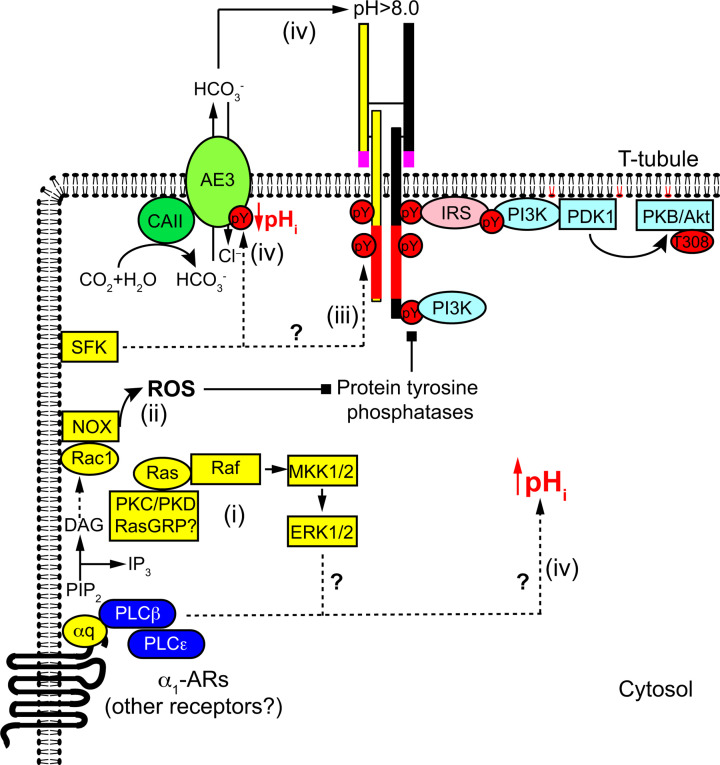
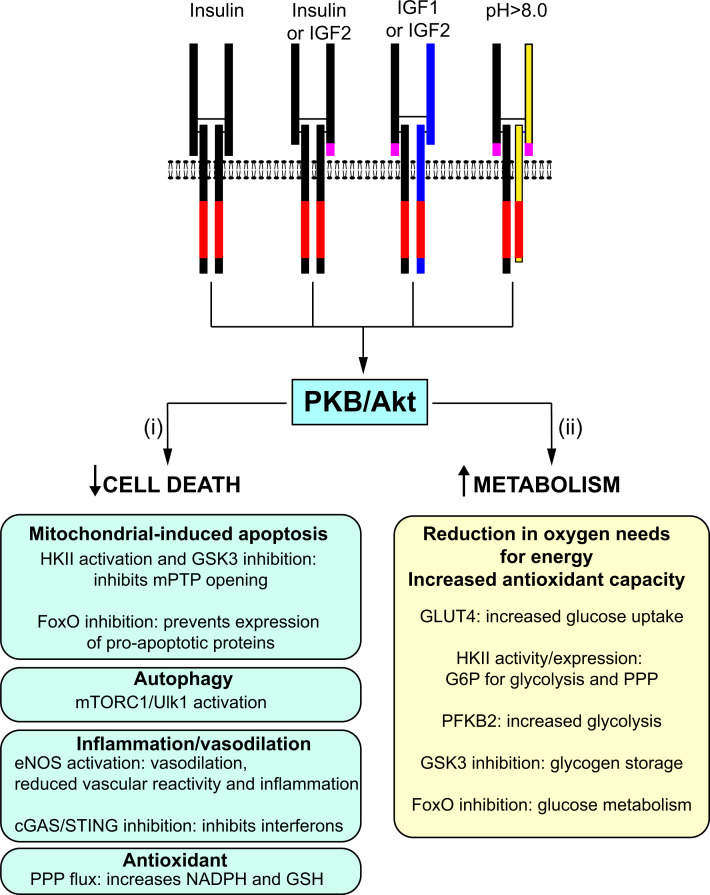
Insulin was discovered over 100 years ago. Whilst the first half century defined many of the physiological effects of insulin, the second emphasised the mechanisms by which it elicits these effects, implicating a vast array of G proteins and their regulators, lipid and protein kinases and counteracting phosphatases, and more. Potential growth-promoting and protective effects of insulin on the heart emerged from studies of carbohydrate metabolism in the 1960s, but the insulin receptors (and the related receptor for insulin-like growth factors 1 and 2) were not defined until the 1980s. A related third receptor, the insulin receptor-related receptor remained an orphan receptor for many years until it was identified as an alkali-sensor. The mechanisms by which these receptors and the plethora of downstream signalling molecules confer cardioprotection remain elusive. Here, we review important aspects of the effects of the three insulin receptor family members in the heart. Metabolic studies are set in the context of what is now known of insulin receptor family signalling and the role of protein kinase B (PKB or Akt), and the relationship between this and cardiomyocyte survival versus death is discussed. PKB/Akt phosphorylates numerous substrates with potential for cardioprotection in the contractile cardiomyocytes and cardiac non-myocytes. Our overall conclusion is that the effects of insulin on glucose metabolism that were initially identified remain highly pertinent in managing cardiomyocyte energetics and preservation of function. This alone provides a high level of cardioprotection in the face of pathophysiological stressors such as ischaemia and myocardial infarction.
Keywords: insulin receptors; insulin signalling; intracellular signaling; regulation of metabolism.
© 2022 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
Similar articles
-
The insulin receptor family and protein kinase B (Akt) are activated in the heart by alkaline pH and α1-adrenergic receptors.Biochem J. 2021 Jun 11;478(11):2059-2079. doi: 10.1042/BCJ20210144. Biochem J. 2021. PMID: 34002209 Free PMC article.
-
Metallothionein antagonizes aging-induced cardiac contractile dysfunction: role of PTP1B, insulin receptor tyrosine phosphorylation and Akt.Aging Cell. 2006 Apr;5(2):177-85. doi: 10.1111/j.1474-9726.2006.00201.x. Aging Cell. 2006. PMID: 16626396
-
FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases.Proc Natl Acad Sci U S A. 2007 Dec 18;104(51):20517-22. doi: 10.1073/pnas.0610290104. Epub 2007 Dec 12. Proc Natl Acad Sci U S A. 2007. PMID: 18077353 Free PMC article.
-
Signalling by insulin and IGF receptors: supporting acts and new players.J Mol Endocrinol. 2011 Jun 17;47(1):R1-10. doi: 10.1530/JME-11-0022. Print 2011 Aug. J Mol Endocrinol. 2011. PMID: 21498522 Review.
-
Regulation of insulin resistance and glucose metabolism by interaction of PIM kinases and insulin receptor substrates.Arch Physiol Biochem. 2020 May;126(2):129-138. doi: 10.1080/13813455.2018.1498903. Epub 2018 Sep 29. Arch Physiol Biochem. 2020. PMID: 30270668 Review.
Cited by
-
Protein-protein interaction network-based integration of GWAS and functional data for blood pressure regulation analysis.Hum Genomics. 2024 Feb 8;18(1):15. doi: 10.1186/s40246-023-00565-6. Hum Genomics. 2024. PMID: 38326862 Free PMC article.
-
Slowing Heart Rate Protects Against Pathological Cardiac Hypertrophy.Function (Oxf). 2022 Nov 1;4(1):zqac055. doi: 10.1093/function/zqac055. eCollection 2023. Function (Oxf). 2022. PMID: 36540889 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous