

Structural biology of ex vivo mammalian prions
- PMID: 35752366
- PMCID: PMC9293645
- DOI: 10.1016/j.jbc.2022.102181
Structural biology of ex vivo mammalian prions
Abstract
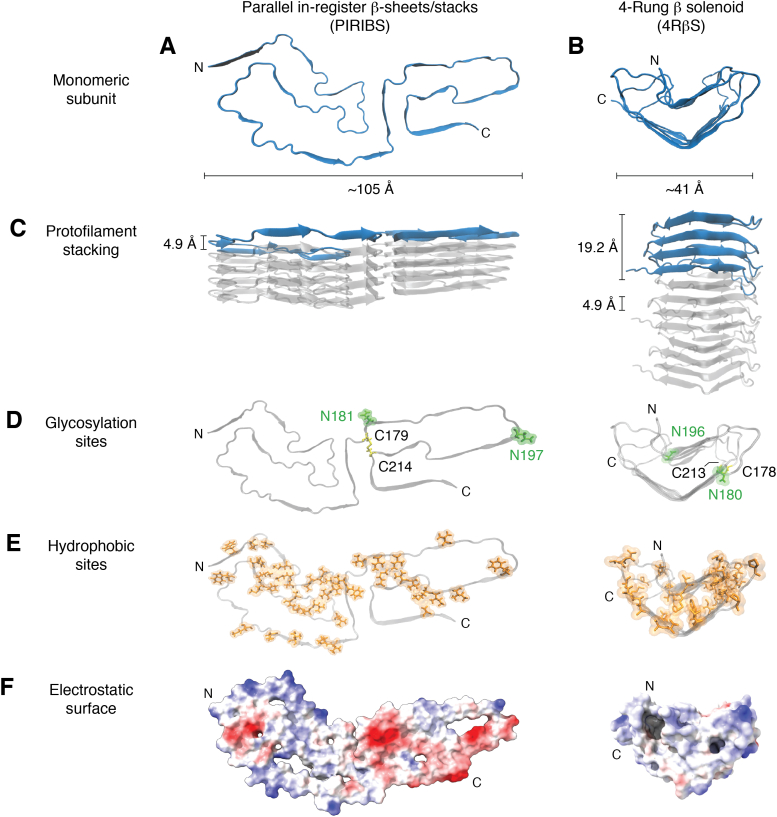
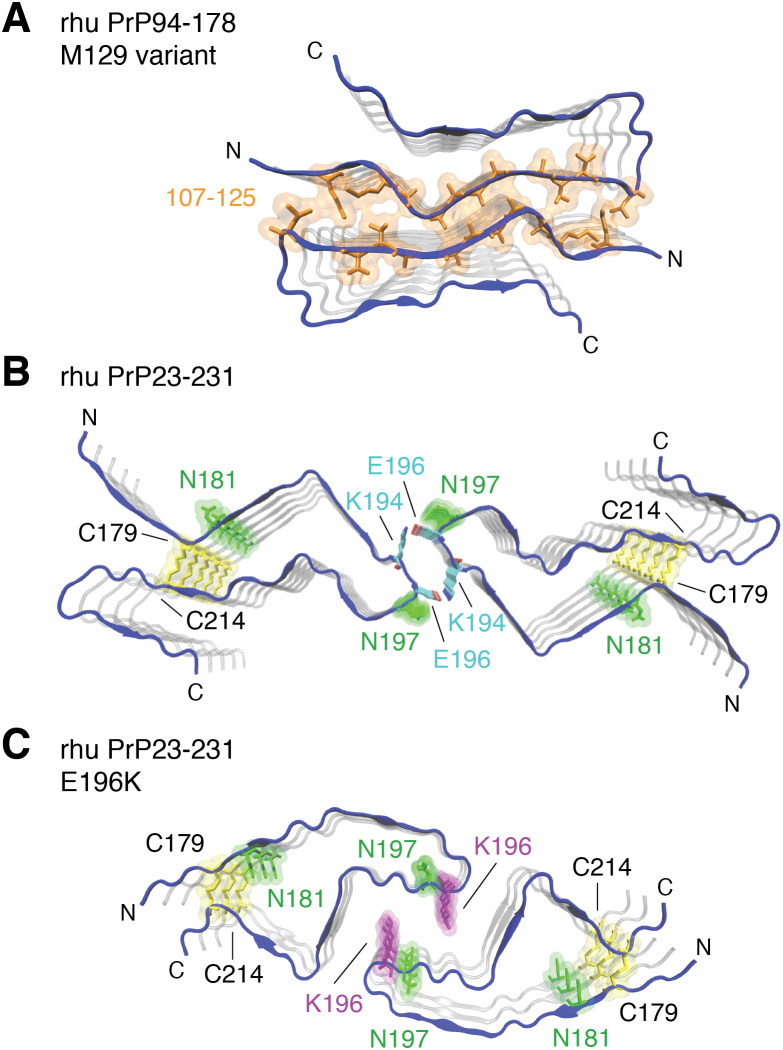
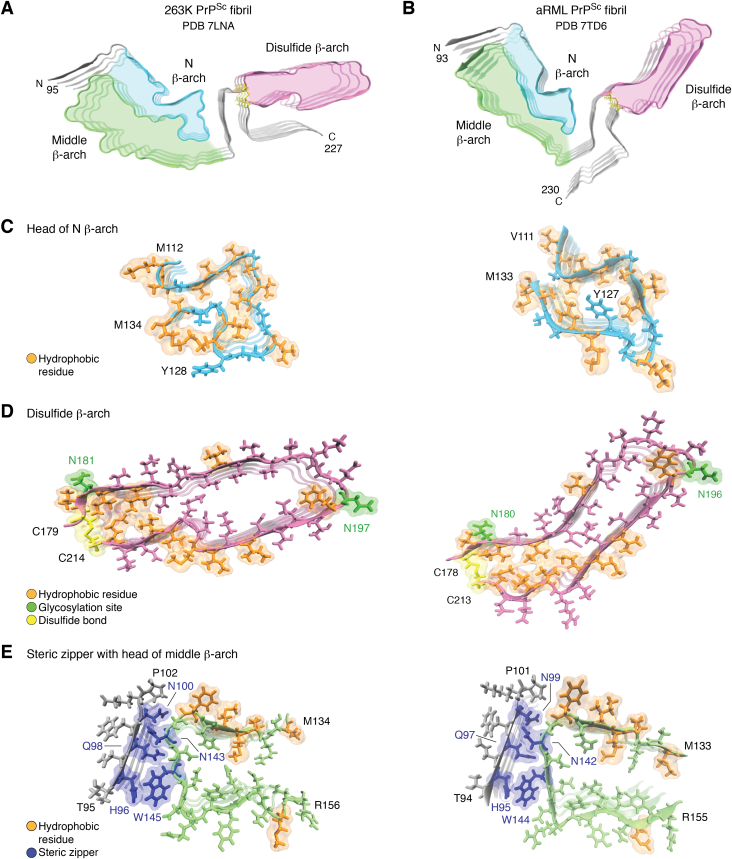
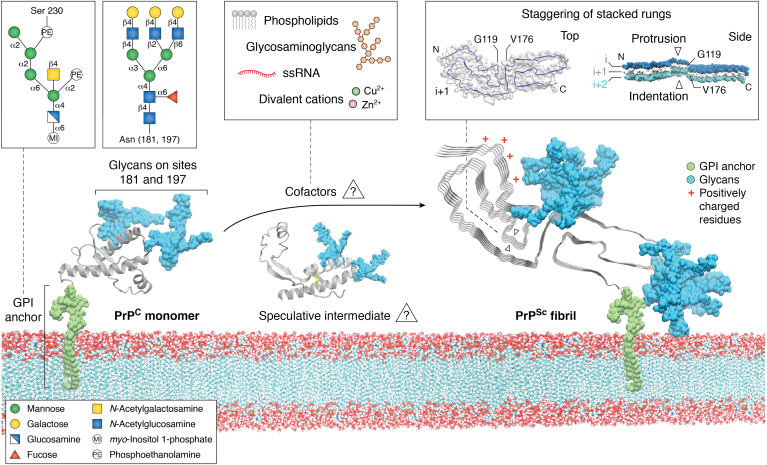
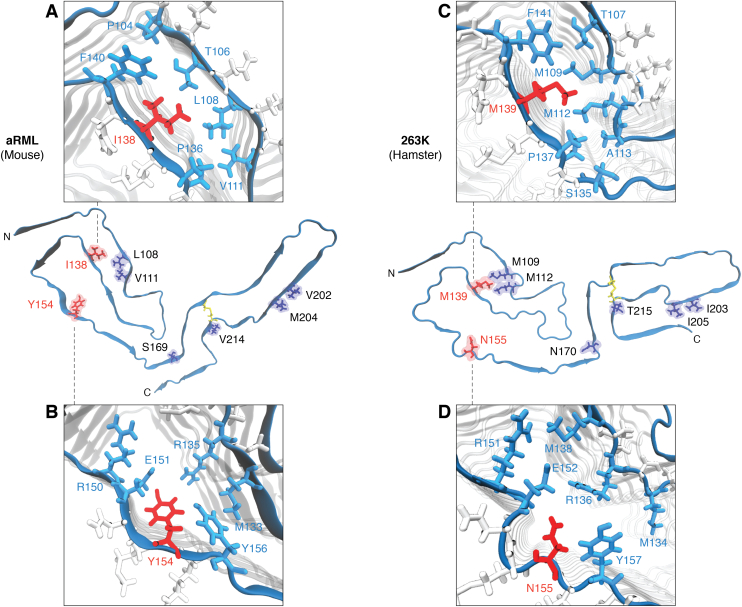
The structures of prion protein (PrP)-based mammalian prions have long been elusive. However, cryo-EM has begun to reveal the near-atomic resolution structures of fully infectious ex vivo mammalian prion fibrils as well as relatively innocuous synthetic PrP amyloids. Comparisons of these various types of PrP fibrils are now providing initial clues to structural features that correlate with pathogenicity. As first indicated by electron paramagnetic resonance and solid-state NMR studies of synthetic amyloids, all sufficiently resolved PrP fibrils of any sort (n > 10) have parallel in-register intermolecular β-stack architectures. Cryo-EM has shown that infectious brain-derived prion fibrils of the rodent-adapted 263K and RML scrapie strains have much larger ordered cores than the synthetic fibrils. These bona fide prion strains share major structural motifs, but the conformational details and the overall shape of the fibril cross sections differ markedly. Such motif variations, as well as differences in sequence within the ordered polypeptide cores, likely contribute to strain-dependent templating. When present, N-linked glycans and glycophosphatidylinositol (GPI) anchors project outward from the fibril surface. For the mouse RML strain, these posttranslational modifications have little effect on the core structure. In the GPI-anchored prion structures, a linear array of GPI anchors along the twisting fibril axis appears likely to bind membranes in vivo, and as such, may account for pathognomonic membrane distortions seen in prion diseases. In this review, we focus on these infectious prion structures and their implications regarding prion replication mechanisms, strains, transmission barriers, and molecular pathogenesis.
Keywords: N-linked glycosylation; amyloid; cryo-EM; glycosylphosphatidylinositol anchor; infectious disease; neurodegenerative disease; prion; prion disease; protein aggregation; protein structure.
Published by Elsevier Inc.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
Cryo-EM structure of anchorless RML prion reveals variations in shared motifs between distinct strains.Nat Commun. 2022 Jul 13;13(1):4005. doi: 10.1038/s41467-022-30458-6. Nat Commun. 2022. PMID: 35831291 Free PMC article.
-
2.7 Å cryo-EM structure of ex vivo RML prion fibrils.Nat Commun. 2022 Jul 13;13(1):4004. doi: 10.1038/s41467-022-30457-7. Nat Commun. 2022. PMID: 35831275 Free PMC article.
-
Prion strains viewed through the lens of cryo-EM.Cell Tissue Res. 2023 Apr;392(1):167-178. doi: 10.1007/s00441-022-03676-z. Epub 2022 Aug 27. Cell Tissue Res. 2023. PMID: 36028585 Free PMC article. Review.
-
High-resolution structure and strain comparison of infectious mammalian prions.Mol Cell. 2021 Nov 4;81(21):4540-4551.e6. doi: 10.1016/j.molcel.2021.08.011. Epub 2021 Aug 25. Mol Cell. 2021. PMID: 34433091
-
Molecular structures of amyloid and prion fibrils: consensus versus controversy.Acc Chem Res. 2013 Jul 16;46(7):1487-96. doi: 10.1021/ar300282r. Epub 2013 Jan 7. Acc Chem Res. 2013. PMID: 23294335 Free PMC article. Review.
Cited by
-
Cryo-EM structure of disease-related prion fibrils provides insights into seeding barriers.Nat Struct Mol Biol. 2022 Oct;29(10):962-965. doi: 10.1038/s41594-022-00833-4. Epub 2022 Sep 12. Nat Struct Mol Biol. 2022. PMID: 36097290 Free PMC article.
-
Effect of host and strain factors on α-synuclein prion pathogenesis.Trends Neurosci. 2024 Jul;47(7):538-550. doi: 10.1016/j.tins.2024.05.004. Epub 2024 May 27. Trends Neurosci. 2024. PMID: 38806297 Review.
-
Sensitive detection of pathological seeds of α-synuclein, tau and prion protein on solid surfaces.PLoS Pathog. 2024 Apr 19;20(4):e1012175. doi: 10.1371/journal.ppat.1012175. eCollection 2024 Apr. PLoS Pathog. 2024. PMID: 38640117 Free PMC article.
-
Protective role of cytosolic prion protein against virus infection in prion-infected cells.J Virol. 2024 Sep 17;98(9):e0126224. doi: 10.1128/jvi.01262-24. Epub 2024 Aug 28. J Virol. 2024. PMID: 39194237
-
Minor prion substrains overcome transmission barriers.mBio. 2024 Nov 13;15(11):e0272124. doi: 10.1128/mbio.02721-24. Epub 2024 Oct 23. mBio. 2024. PMID: 39440977 Free PMC article.
References
-
- Jeffrey M., McGovern G., Siso S., Gonzalez L. Cellular and sub-cellular pathology of animal prion diseases: Relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011;121:113–134. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
