

Cathepsin B Gene Knockout Improves Behavioral Deficits and Reduces Pathology in Models of Neurologic Disorders
- PMID: 35710131
- PMCID: PMC9553114
- DOI: 10.1124/pharmrev.121.000527
Cathepsin B Gene Knockout Improves Behavioral Deficits and Reduces Pathology in Models of Neurologic Disorders
Abstract
Cathepsin B (CTSB) is a powerful lysosomal protease. This review evaluated CTSB gene knockout (KO) outcomes for amelioration of brain dysfunctions in neurologic diseases and aging animal models. Deletion of the CTSB gene resulted in significant improvements in behavioral deficits, neuropathology, and/or biomarkers in traumatic brain injury, ischemia, inflammatory pain, opiate tolerance, epilepsy, aging, transgenic Alzheimer's disease (AD), and periodontitis AD models as shown in 12 studies. One study found beneficial effects for double CTSB and cathepsin S KO mice in a multiple sclerosis model. Transgenic AD models using amyloid precursor protein (APP) mimicking common sporadic AD in three studies showed that CTSB KO improved memory, neuropathology, and biomarkers; two studies used APP representing rare familial AD and found no CTSB KO effect, and two studies used highly engineered APP constructs and reported slight increases in a biomarker. In clinical studies, all reports found that CTSB enzyme was upregulated in diverse neurologic disorders, including AD in which elevated CTSB was positively correlated with cognitive dysfunction. In a wide range of neurologic animal models, CTSB was also upregulated and not downregulated. Further, human genetic mutation data provided precedence for CTSB upregulation causing disease. Thus, the consilience of data is that CTSB gene KO results in improved brain dysfunction and reduced pathology through blockade of CTSB enzyme upregulation that causes human neurologic disease phenotypes. The overall findings provide strong support for CTSB as a rational drug target and for CTSB inhibitors as therapeutic candidates for a wide range of neurologic disorders. SIGNIFICANCE STATEMENT: This review provides a comprehensive compilation of the extensive data on the effects of deleting the cathepsin B (CTSB) gene in neurological and aging mouse models of brain disorders. Mice lacking the CTSB gene display improved neurobehavioral deficits, reduced neuropathology, and amelioration of neuronal cell death and inflammatory biomarkers. The significance of the compelling CTSB evidence is that the data consilience validates CTSB as a drug target for discovery of CTSB inhibitors as potential therapeutics for treating numerous neurological diseases.
U.S. Government work not protected by U.S. copyright.
Conflict of interest statement
V.H. and G.H. have equity positions at American Life Science Pharmaceuticals (ALSP) and are founders of ALSP. V.H. is an advisor to ALSP. G.H. is vice president of research, corporate counsel, and member of the board of directors at ALSP. V.H.’s conflict has been disclosed and is managed by her employer, the University of California, San Diego. No other author has an actual or perceived conflict of interest with the contents of this article.
Figures
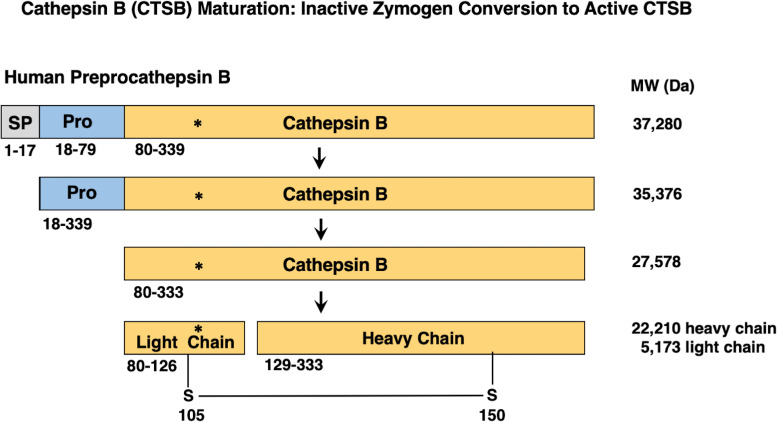
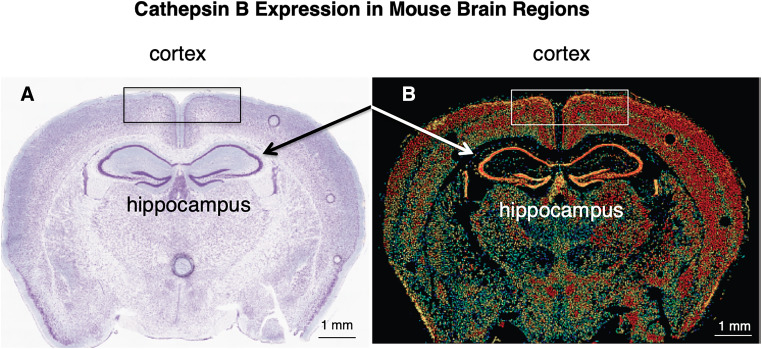

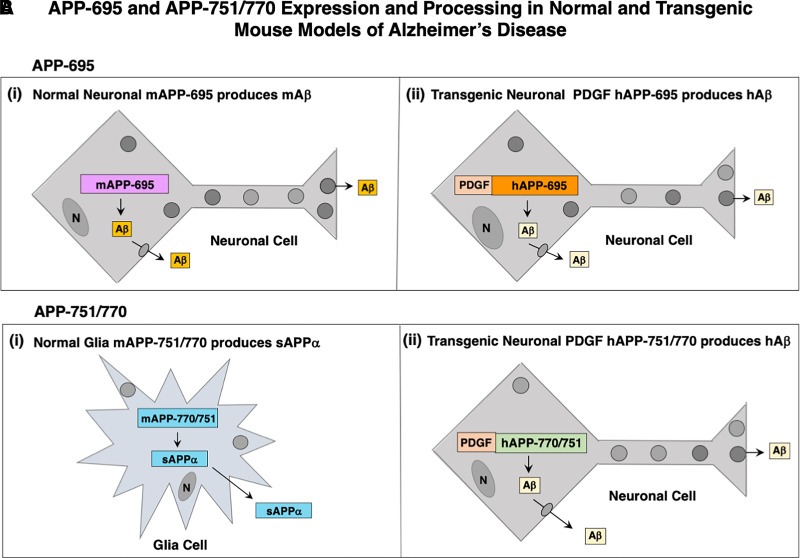

Similar articles
-
Cathepsin B in neurodegeneration of Alzheimer's disease, traumatic brain injury, and related brain disorders.Biochim Biophys Acta Proteins Proteom. 2020 Aug;1868(8):140428. doi: 10.1016/j.bbapap.2020.140428. Epub 2020 Apr 17. Biochim Biophys Acta Proteins Proteom. 2020. PMID: 32305689 Free PMC article. Review.
-
Deletion of the cathepsin B gene improves memory deficits in a transgenic ALZHeimer's disease mouse model expressing AβPP containing the wild-type β-secretase site sequence.J Alzheimers Dis. 2012;29(4):827-40. doi: 10.3233/JAD-2012-111604. J Alzheimers Dis. 2012. PMID: 22337825 Free PMC article.
-
Loss of Cathepsin B and L Leads to Lysosomal Dysfunction, NPC-Like Cholesterol Sequestration and Accumulation of the Key Alzheimer's Proteins.PLoS One. 2016 Nov 30;11(11):e0167428. doi: 10.1371/journal.pone.0167428. eCollection 2016. PLoS One. 2016. PMID: 27902765 Free PMC article.
-
Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer's disease therapeutic.J Alzheimers Dis. 2014;41(1):129-49. doi: 10.3233/JAD-131370. J Alzheimers Dis. 2014. PMID: 24595198 Free PMC article.
-
Pharmacogenetic features of cathepsin B inhibitors that improve memory deficit and reduce beta-amyloid related to Alzheimer's disease.Biol Chem. 2010 Aug;391(8):861-72. doi: 10.1515/BC.2010.110. Biol Chem. 2010. PMID: 20536395 Free PMC article. Review.
Cited by
-
Cathepsin B promotes Aβ proteotoxicity by modulating aging regulating mechanisms.Nat Commun. 2024 Oct 3;15(1):8564. doi: 10.1038/s41467-024-52540-x. Nat Commun. 2024. PMID: 39362844 Free PMC article.
-
Aerobic exercise, an effective prevention and treatment for mild cognitive impairment.Front Aging Neurosci. 2023 Aug 8;15:1194559. doi: 10.3389/fnagi.2023.1194559. eCollection 2023. Front Aging Neurosci. 2023. PMID: 37614470 Free PMC article. Review.
-
Cystatin C Attenuates Perihematomal Secondary Brain Injury by Inhibiting the Cathepsin B/NLRP3 Signaling Pathway in a Rat Model of Intracerebral Hemorrhage.Mol Neurobiol. 2024 Nov;61(11):9646-9662. doi: 10.1007/s12035-024-04195-4. Epub 2024 Apr 27. Mol Neurobiol. 2024. PMID: 38676809
-
Milk Fat Globule Membrane-Containing Protein Powder Promotes Fitness in Caenorhabditis elegans.Nutrients. 2024 Jul 17;16(14):2290. doi: 10.3390/nu16142290. Nutrients. 2024. PMID: 39064733 Free PMC article.
-
Cathepsin B Deficiency Improves Memory Deficits and Reduces Amyloid-β in hAβPP Mouse Models Representing the Major Sporadic Alzheimer's Disease Condition.J Alzheimers Dis. 2023;93(1):33-46. doi: 10.3233/JAD-221005. J Alzheimers Dis. 2023. PMID: 36970896 Free PMC article. Review.
References
-
- Abrahamson M (1994). Cystatins. Methods Enzymol 244:685.–700. - PubMed
-
- Abrahamson M, Barrett AJ, Salvesen G, Grubb A (1986) Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J Biol Chem 261:11282–11289. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
