

Protein folding in vitro and in the cell: From a solitary journey to a team effort
- PMID: 35667131
- PMCID: PMC9636488
- DOI: 10.1016/j.bpc.2022.106821
Protein folding in vitro and in the cell: From a solitary journey to a team effort
Abstract
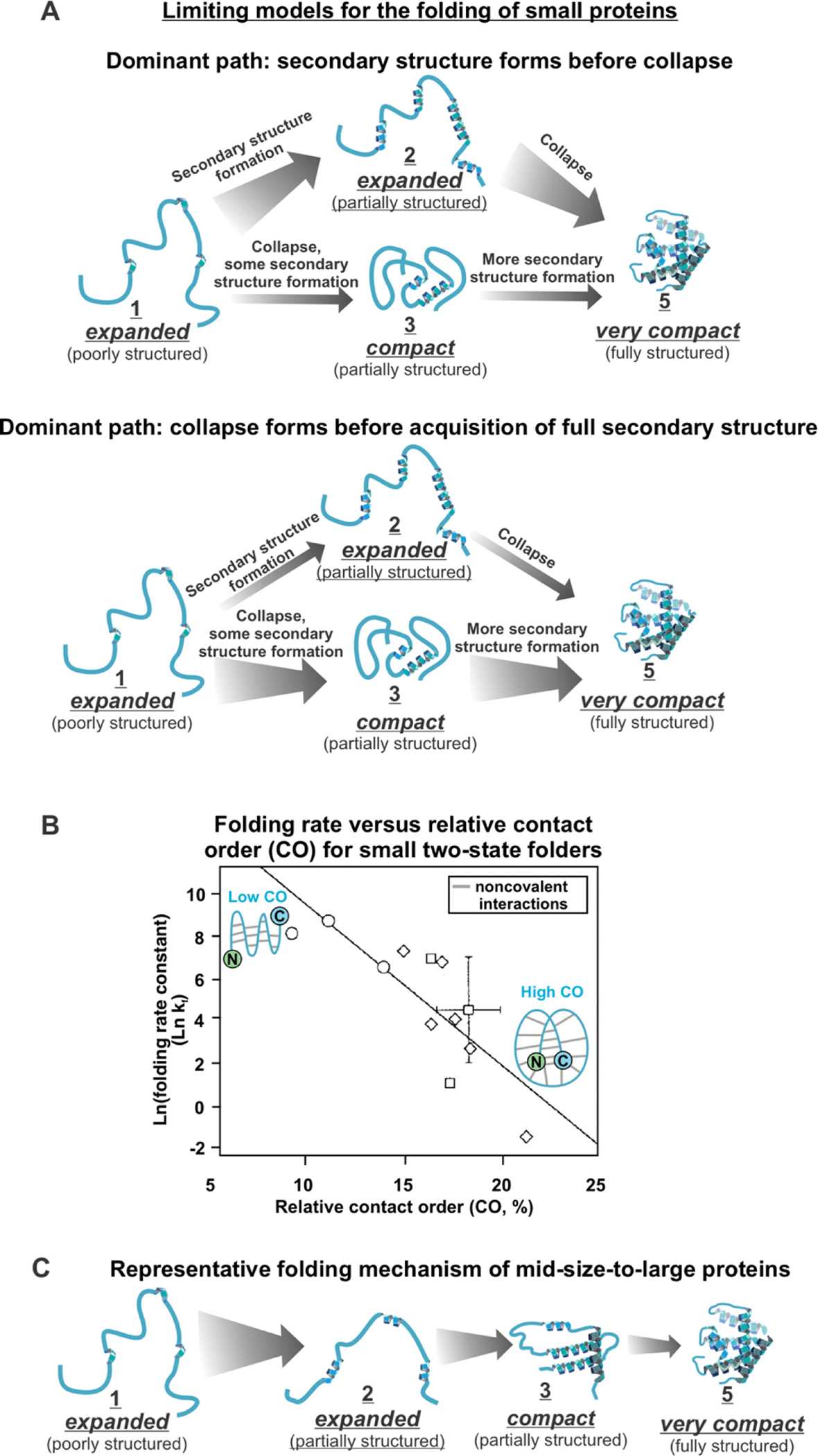
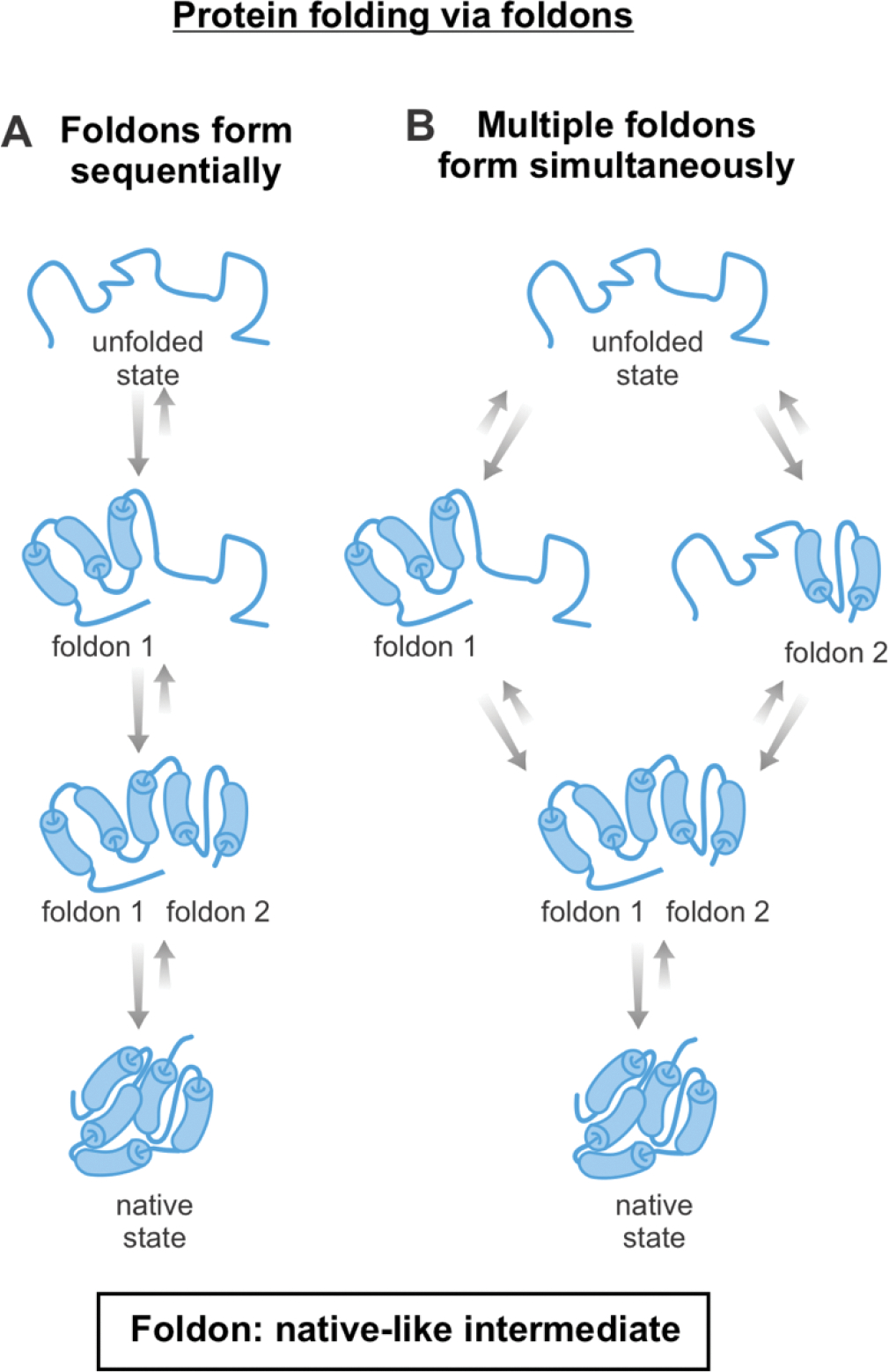
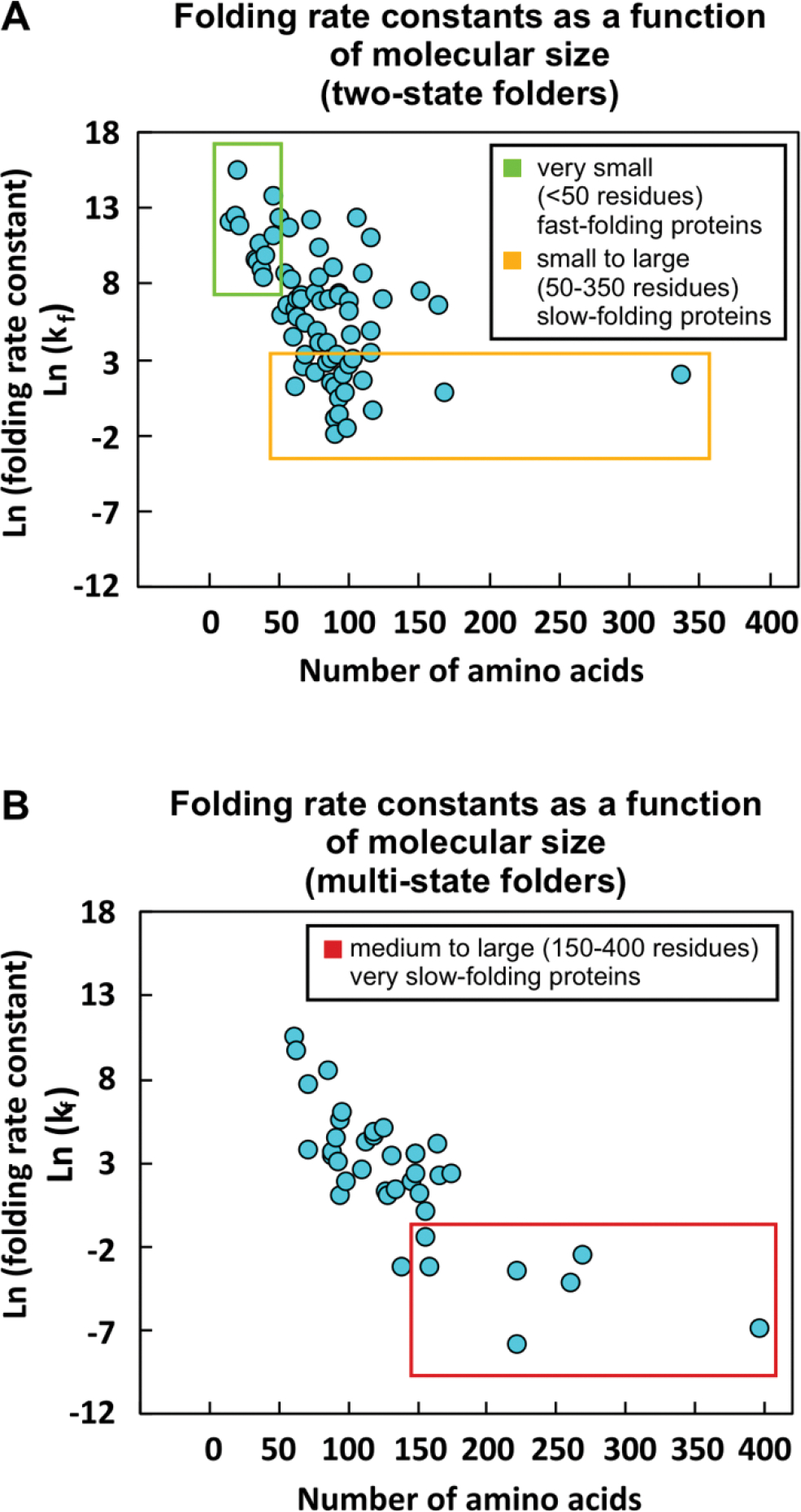
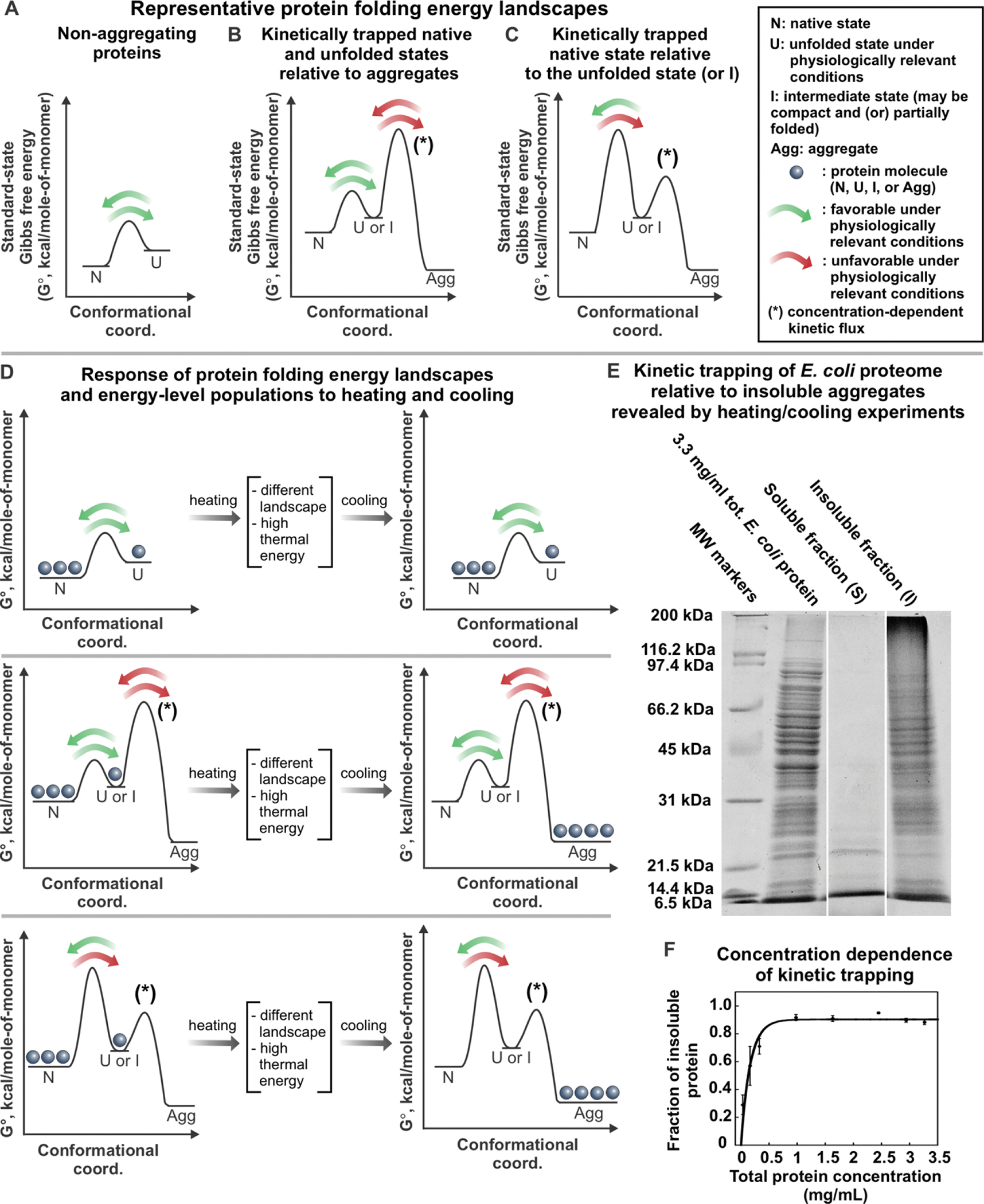
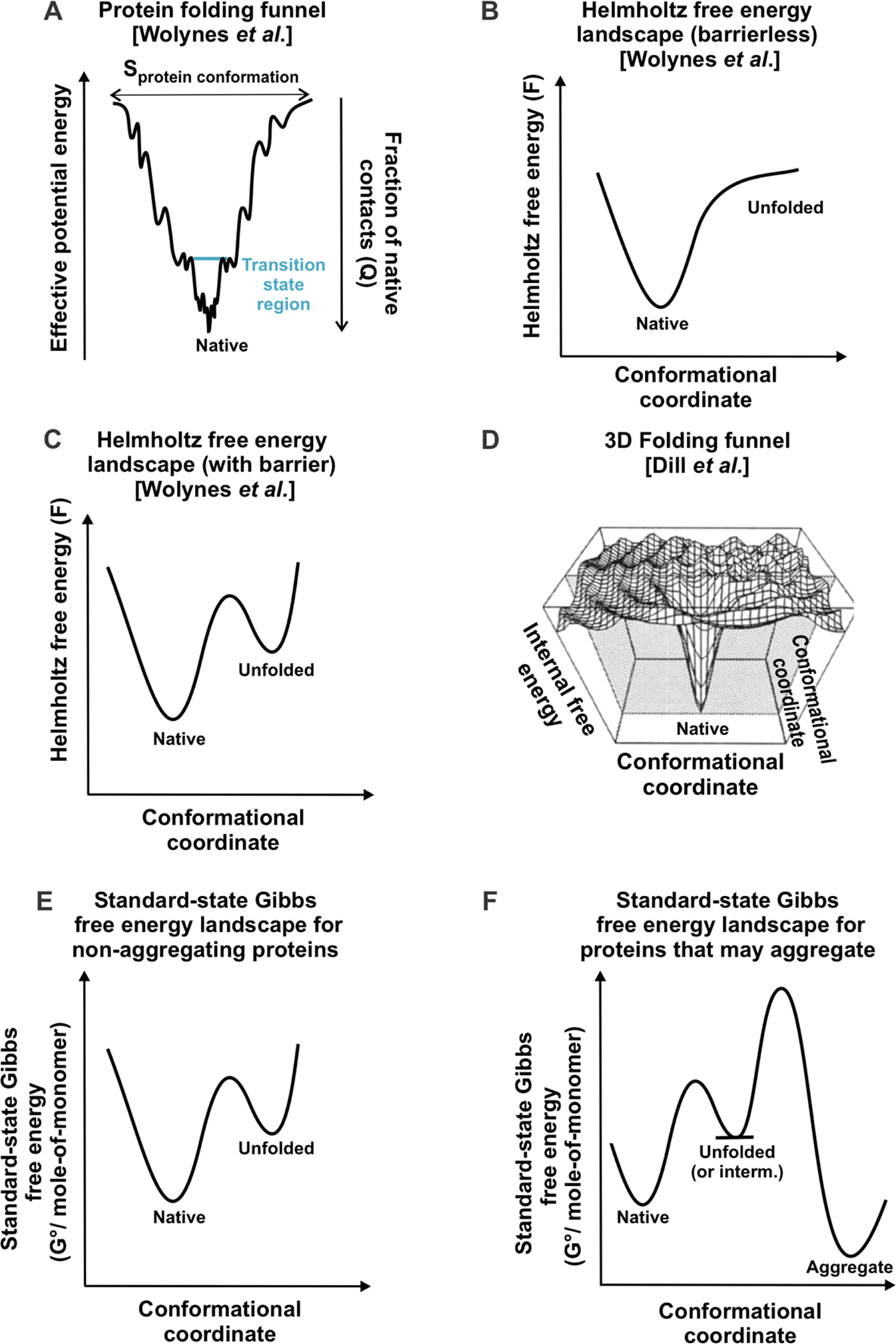
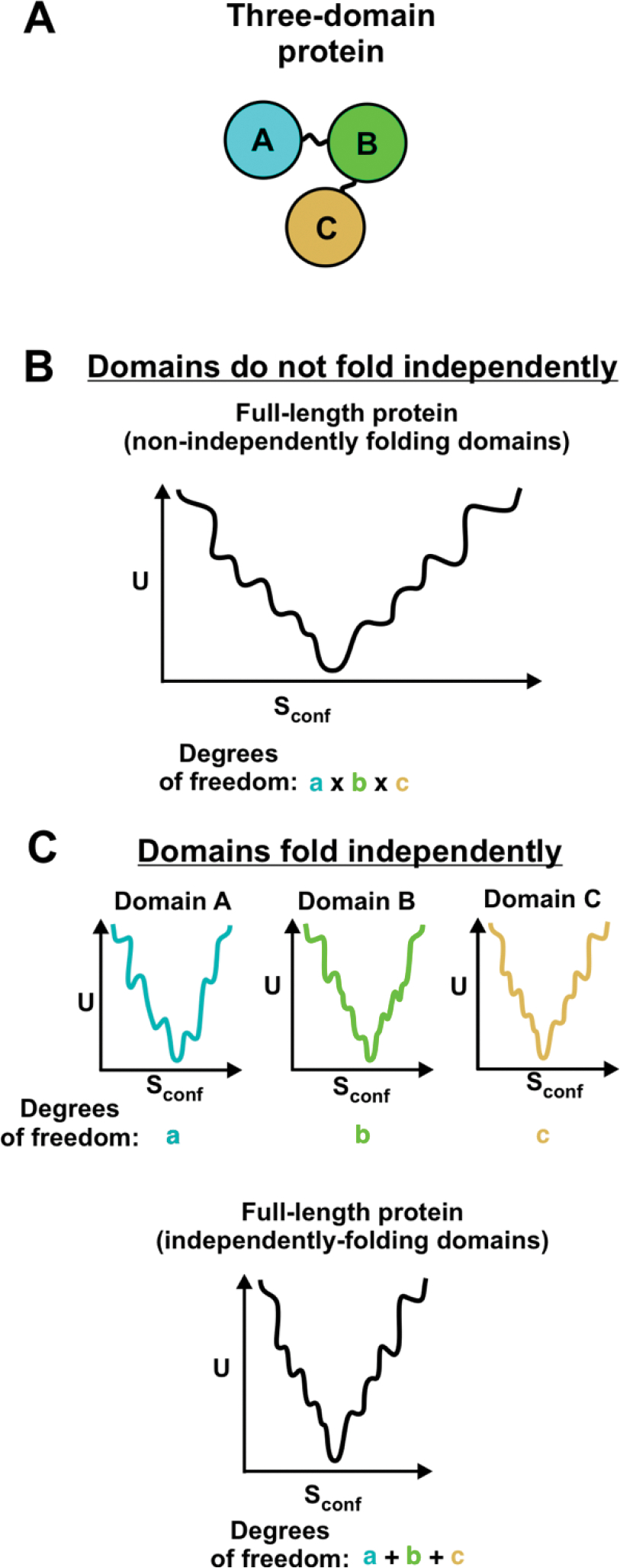
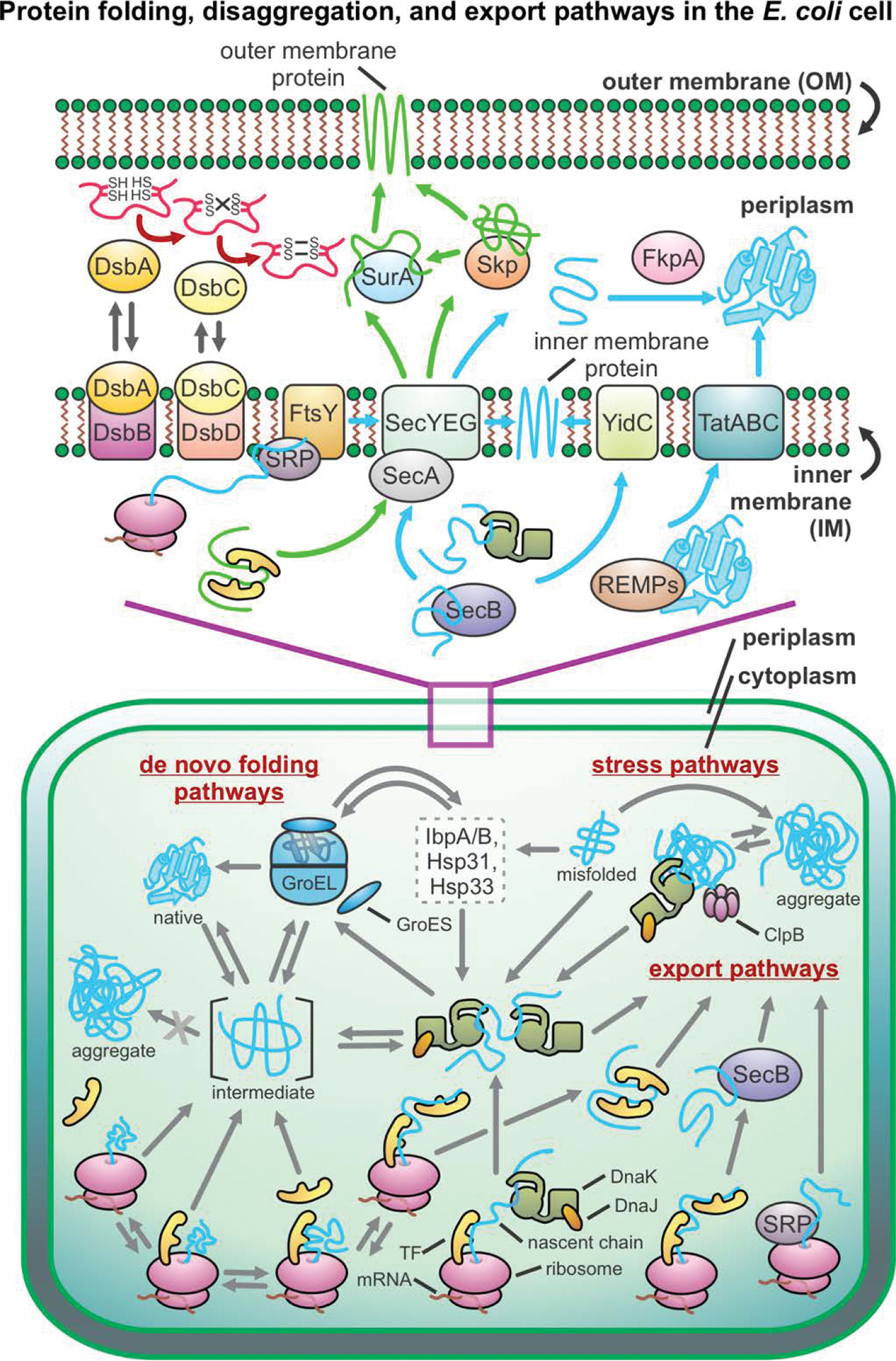
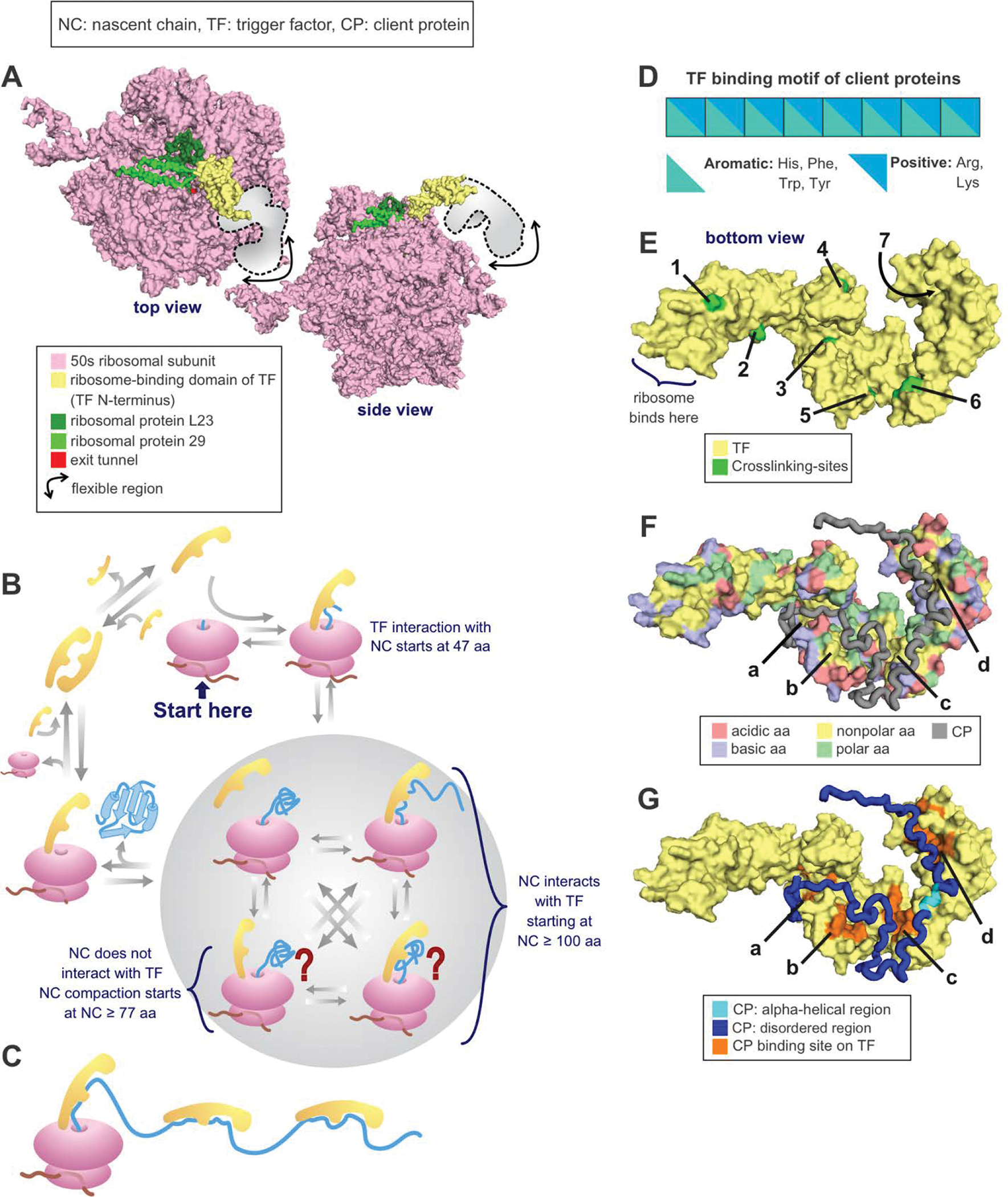
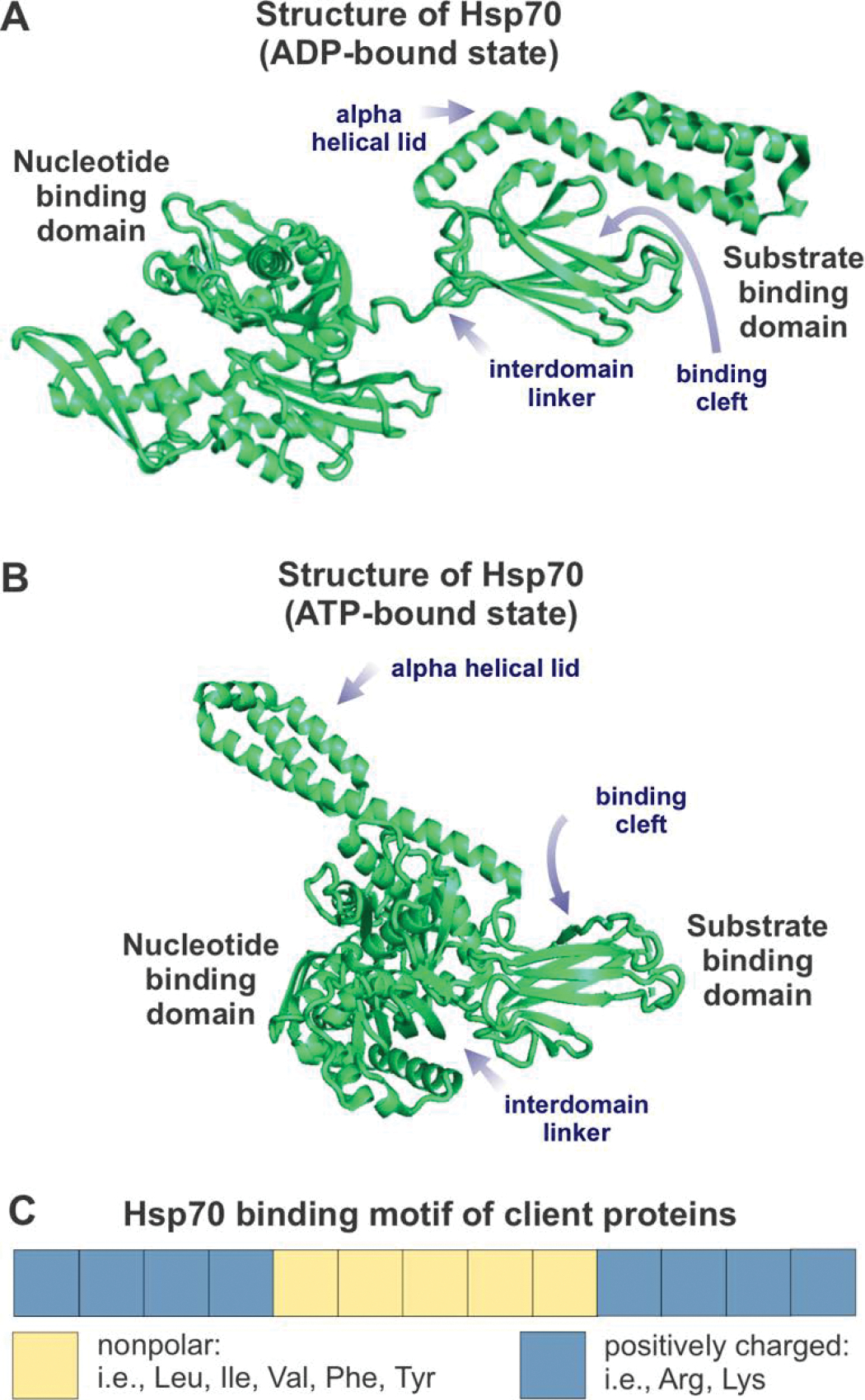
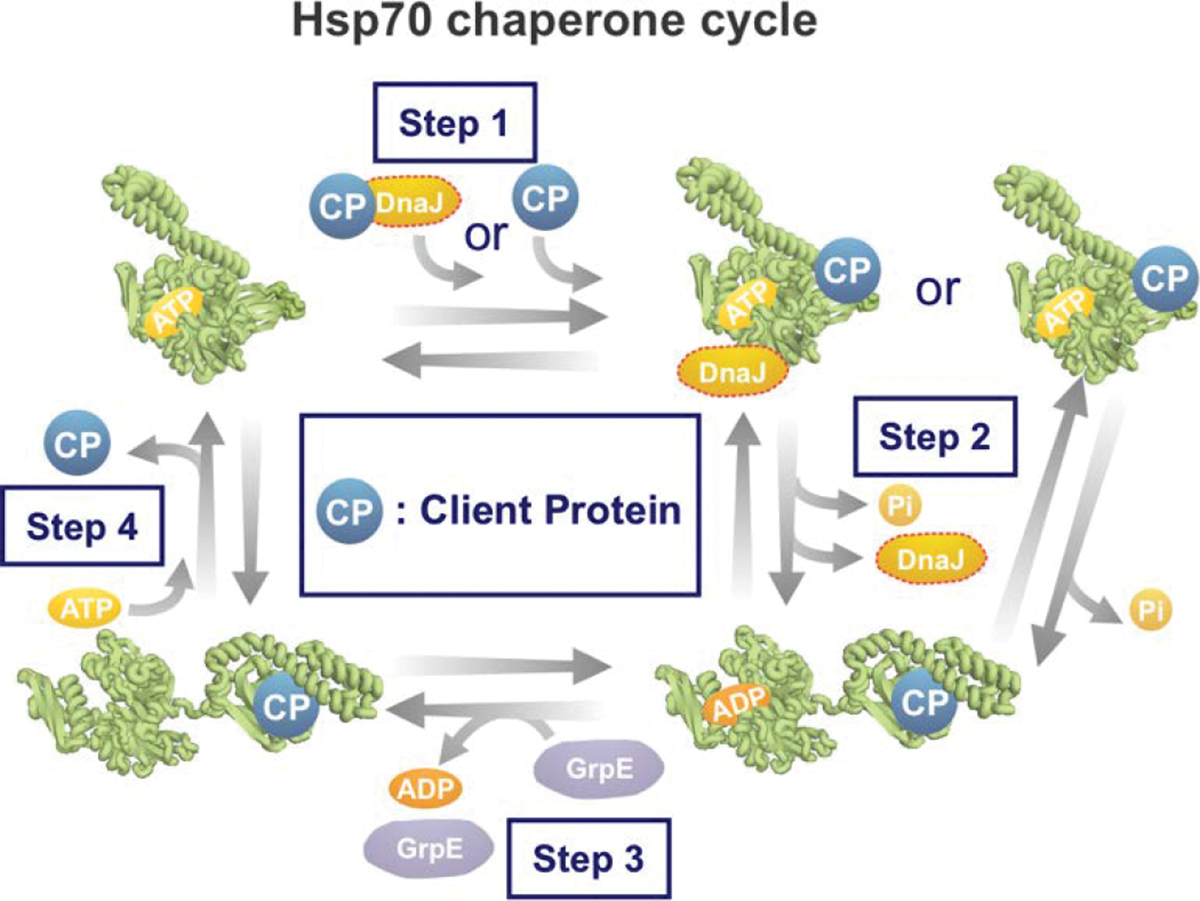
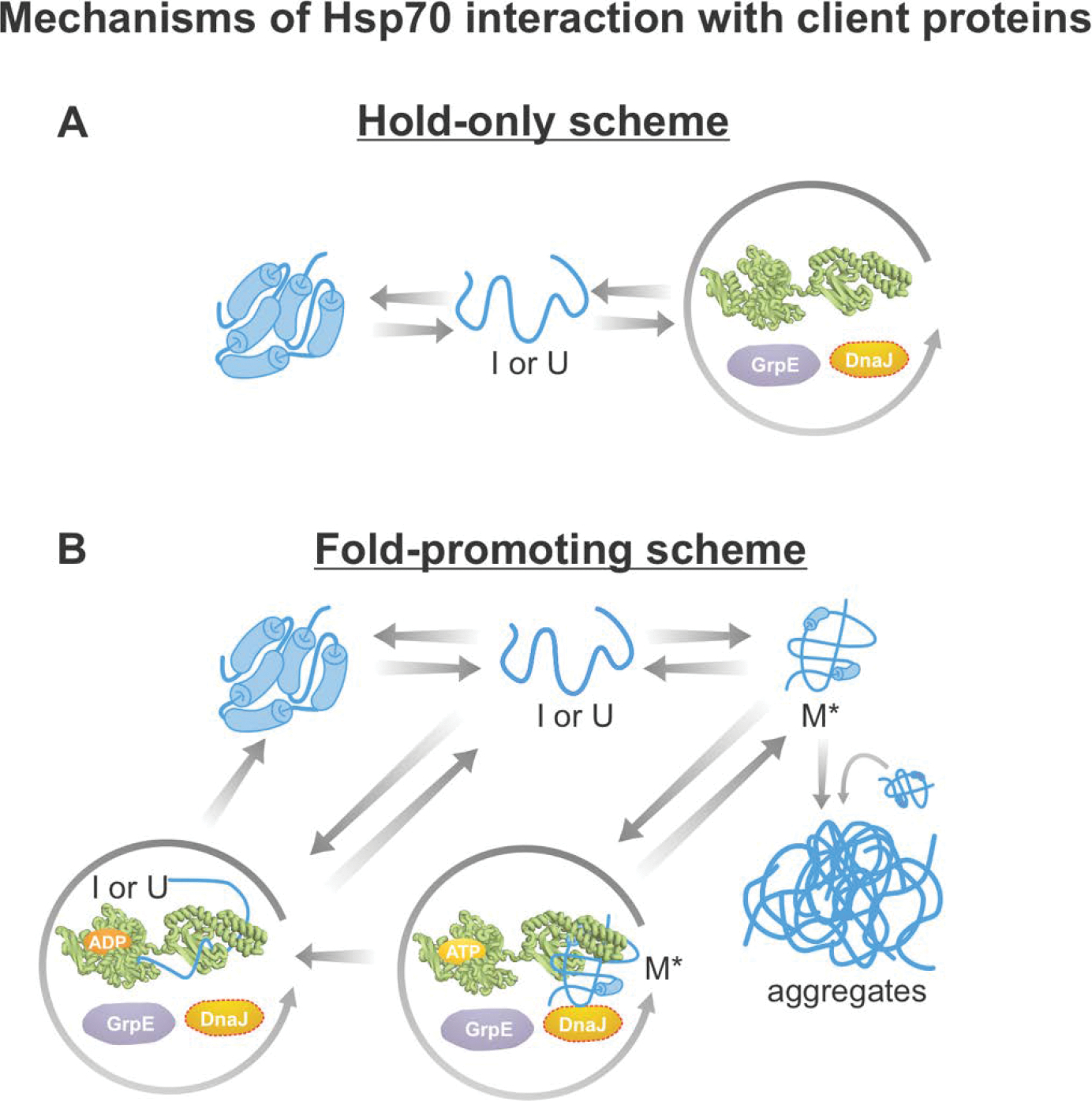
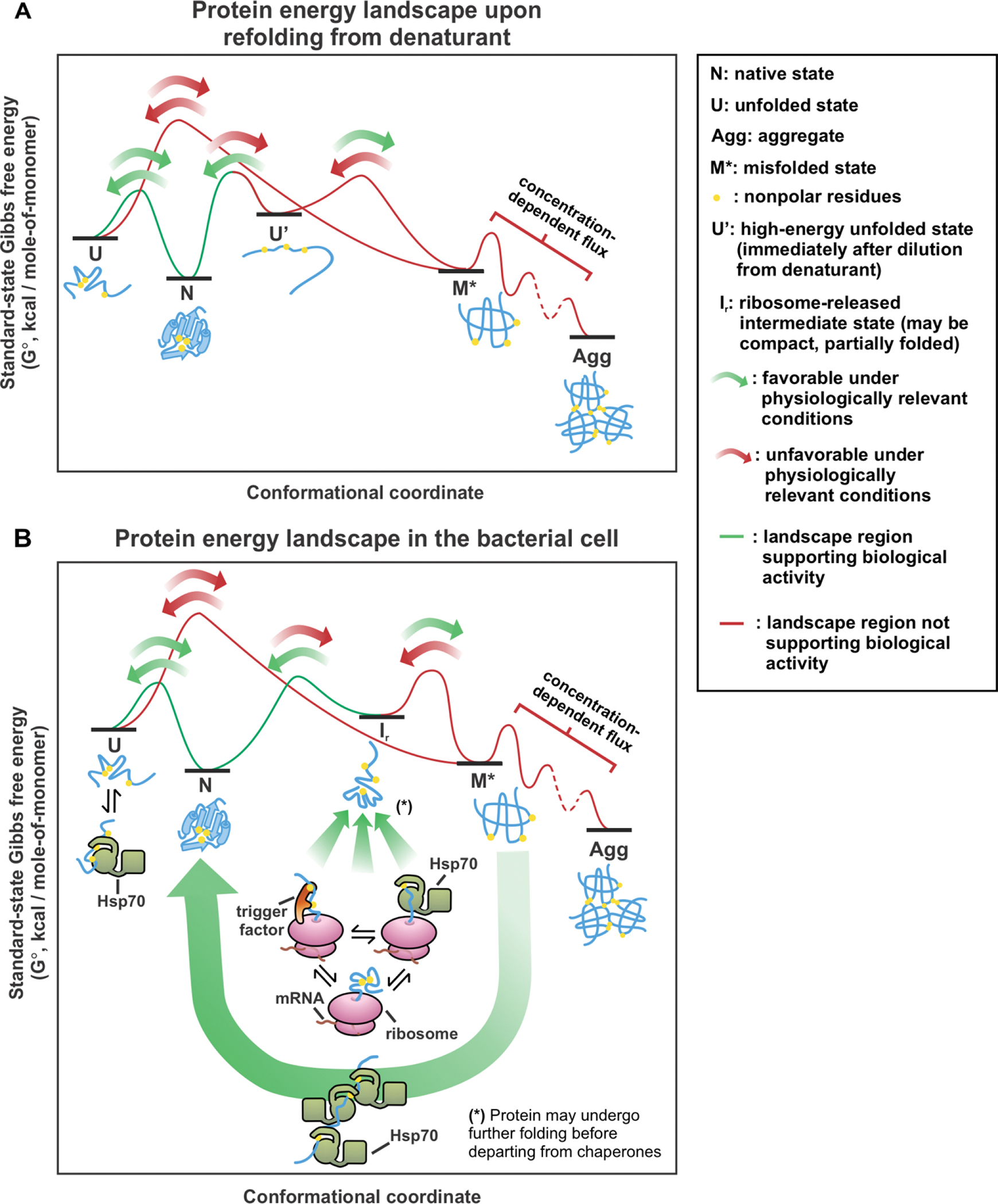
Correct protein folding is essential for the health and function of living organisms. Yet, it is not well understood how unfolded proteins reach their native state and avoid aggregation, especially within the cellular milieu. Some proteins, especially small, single-domain and apparent two-state folders, successfully attain their native state upon dilution from denaturant. Yet, many more proteins undergo misfolding and aggregation during this process, in a concentration-dependent fashion. Once formed, native and aggregated states are often kinetically trapped relative to each other. Hence, the early stages of protein life are absolutely critical for proper kinetic channeling to the folded state and for long-term solubility and function. This review summarizes current knowledge on protein folding/aggregation mechanisms in buffered solution and within the bacterial cell, highlighting early stages. Remarkably, teamwork between nascent chain, ribosome, trigger factor and Hsp70 molecular chaperones enables all proteins to overcome aggregation propensities and reach a long-lived bioactive state.
Keywords: Aggregation; Chaperones; Cotranslational folding; Energy landscapes; Kinetic trapping; Ribosome.
Copyright © 2022 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Complementary Role of Co- and Post-Translational Events in De Novo Protein Biogenesis.J Phys Chem B. 2020 Jul 30;124(30):6488-6507. doi: 10.1021/acs.jpcb.0c03039. Epub 2020 Jun 9. J Phys Chem B. 2020. PMID: 32456434
-
Cotranslational folding increases GFP folding yield.Biophys J. 2010 Apr 7;98(7):1312-20. doi: 10.1016/j.bpj.2009.12.4291. Biophys J. 2010. PMID: 20371331 Free PMC article.
-
Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo.J Mol Biol. 2008 Nov 14;383(3):683-92. doi: 10.1016/j.jmb.2008.07.035. Epub 2008 Jul 22. J Mol Biol. 2008. PMID: 18674543 Free PMC article.
-
Protein folding by NMR.Prog Nucl Magn Reson Spectrosc. 2017 May;100:52-77. doi: 10.1016/j.pnmrs.2016.10.002. Epub 2016 Nov 9. Prog Nucl Magn Reson Spectrosc. 2017. PMID: 28552172 Review.
-
Cotranslational Folding of Proteins on the Ribosome.Biomolecules. 2020 Jan 7;10(1):97. doi: 10.3390/biom10010097. Biomolecules. 2020. PMID: 31936054 Free PMC article. Review.
Cited by
-
Exosomal misfolded proteins released by cancer stem cells: dual functions in balancing protein homeostasis and orchestrating tumor progression.Discov Oncol. 2024 Aug 31;15(1):392. doi: 10.1007/s12672-024-01262-z. Discov Oncol. 2024. PMID: 39215782 Free PMC article. Review.
-
Mapping Protein-Protein Interactions at Birth: Single-Particle Cryo-EM Analysis of a Ribosome-Nascent Globin Complex.ACS Cent Sci. 2024 Feb 1;10(2):385-401. doi: 10.1021/acscentsci.3c00777. eCollection 2024 Feb 28. ACS Cent Sci. 2024. PMID: 38435509 Free PMC article.
-
Nascent chains derived from a foldable protein sequence interact with specific ribosomal surface sites near the exit tunnel.Sci Rep. 2024 May 29;14(1):12324. doi: 10.1038/s41598-024-61274-1. Sci Rep. 2024. PMID: 38811604 Free PMC article.
-
Is It Still Possible to Think about HSP70 as a Therapeutic Target in Onco-Hematological Diseases?Biomolecules. 2023 Mar 28;13(4):604. doi: 10.3390/biom13040604. Biomolecules. 2023. PMID: 37189352 Free PMC article. Review.
-
Distribution and solvent exposure of Hsp70 chaperone binding sites across the Escherichia coli proteome.Proteins. 2023 May;91(5):665-678. doi: 10.1002/prot.26456. Epub 2023 Jan 1. Proteins. 2023. PMID: 36539330 Free PMC article.
References
-
- Speed MA, Wang DI, King J, Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition, Nat. Biotechnol. 14 (1996) 1283–1287. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
