

COVID-19-associated Lung Microvascular Endotheliopathy: A "From the Bench" Perspective
- PMID: 35649173
- PMCID: PMC9801996
- DOI: 10.1164/rccm.202107-1774OC
COVID-19-associated Lung Microvascular Endotheliopathy: A "From the Bench" Perspective
Abstract
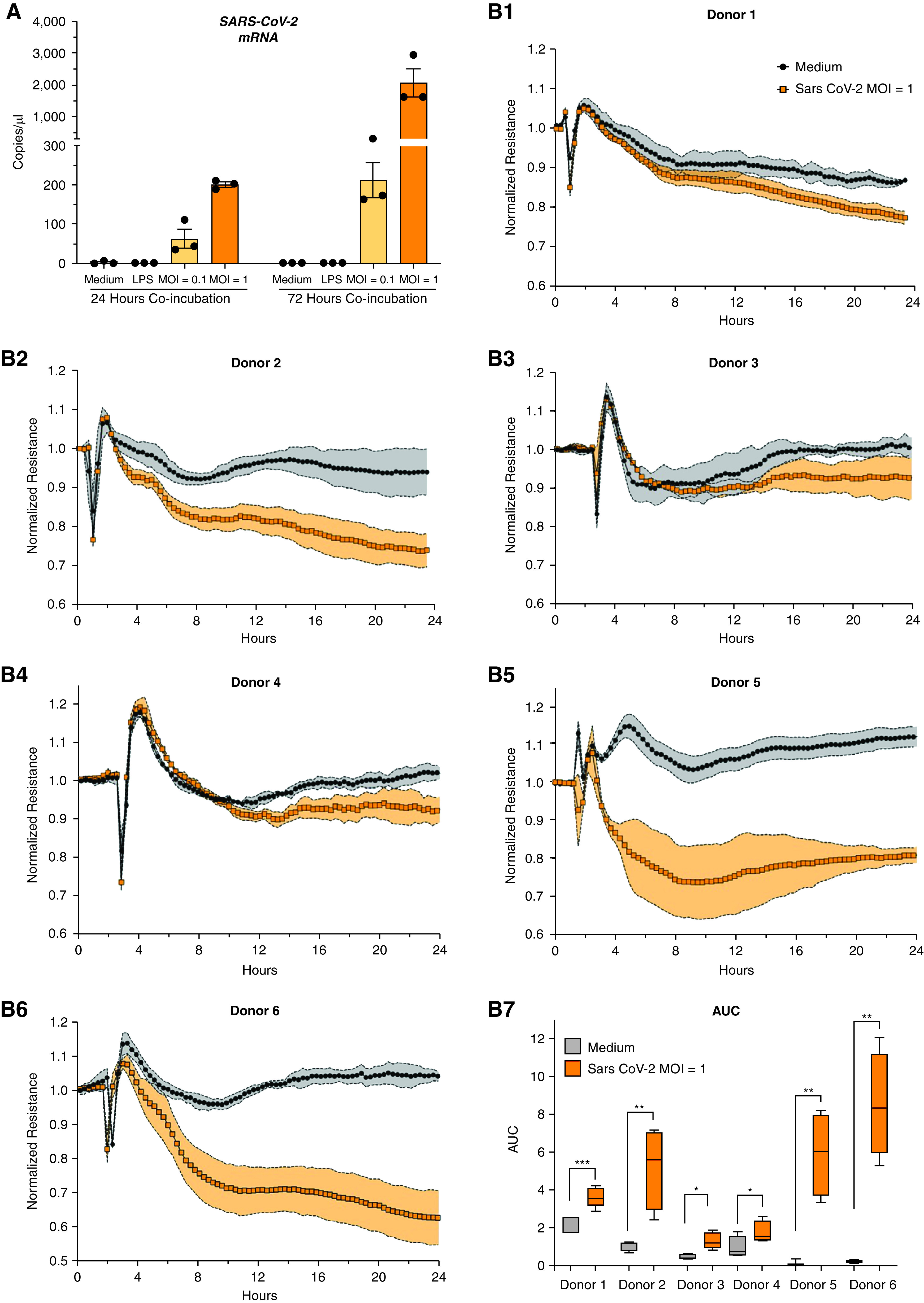
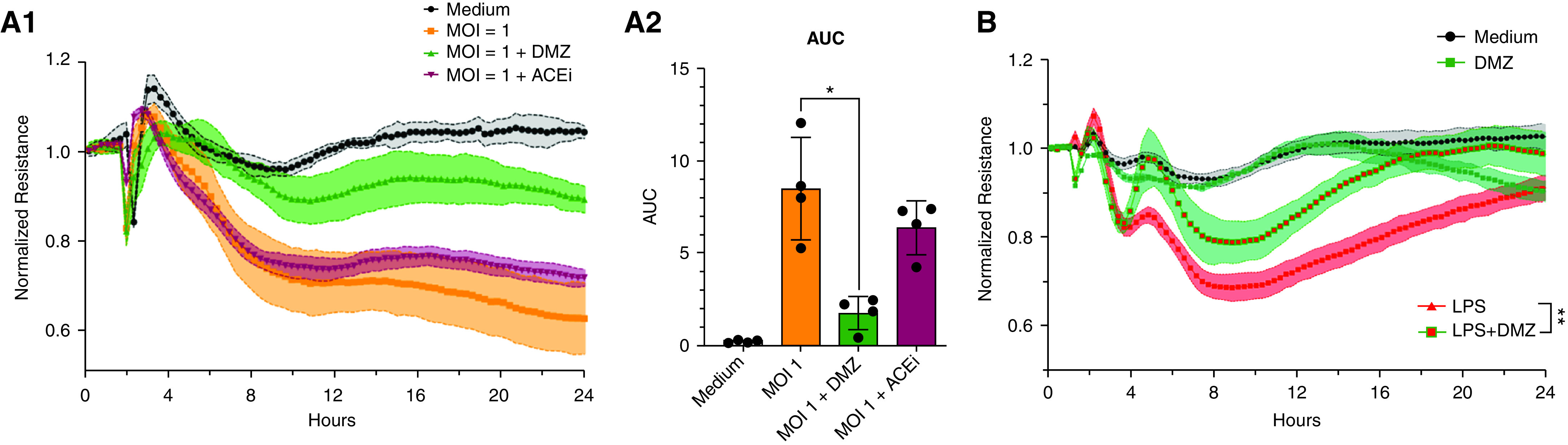
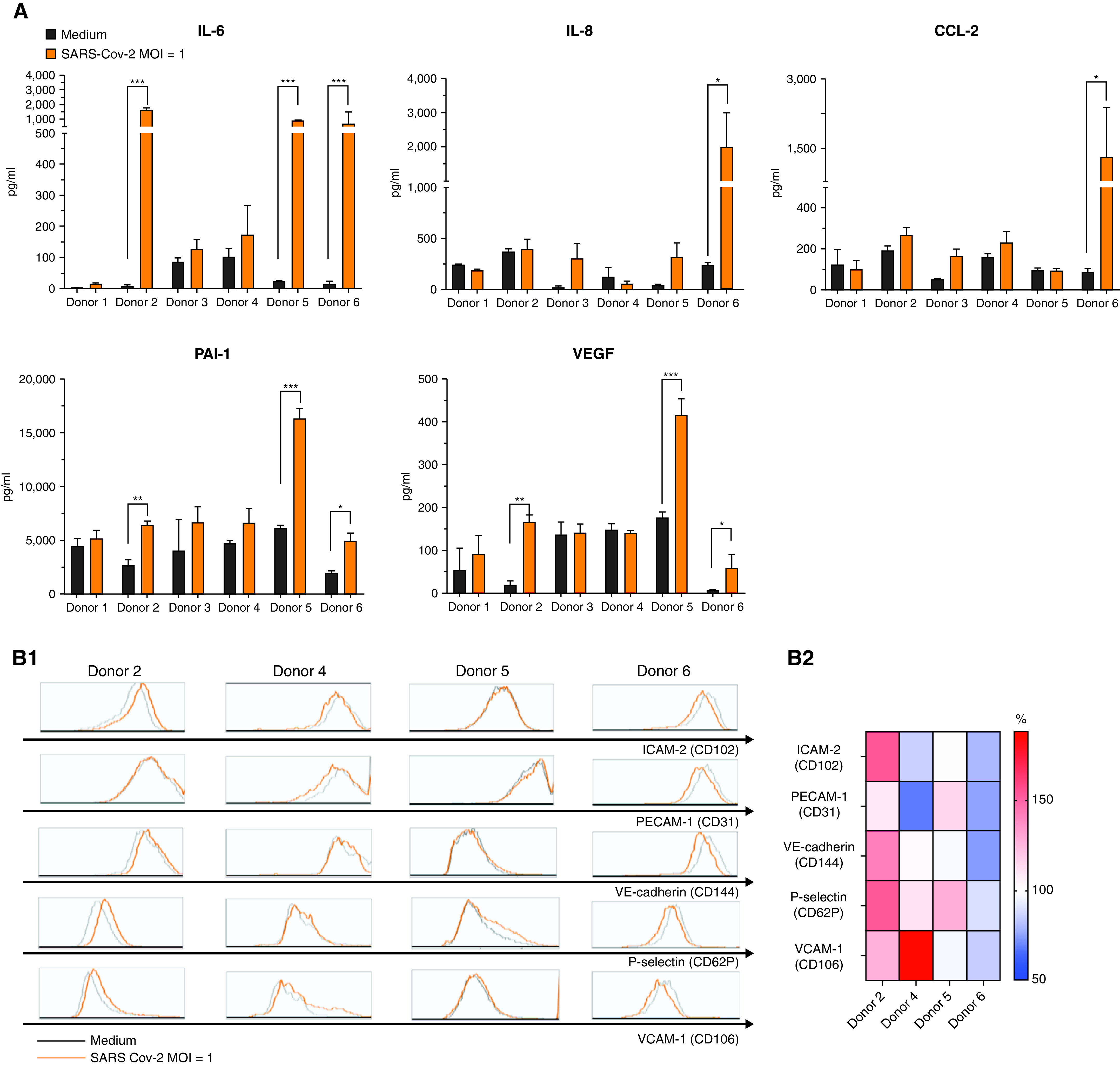
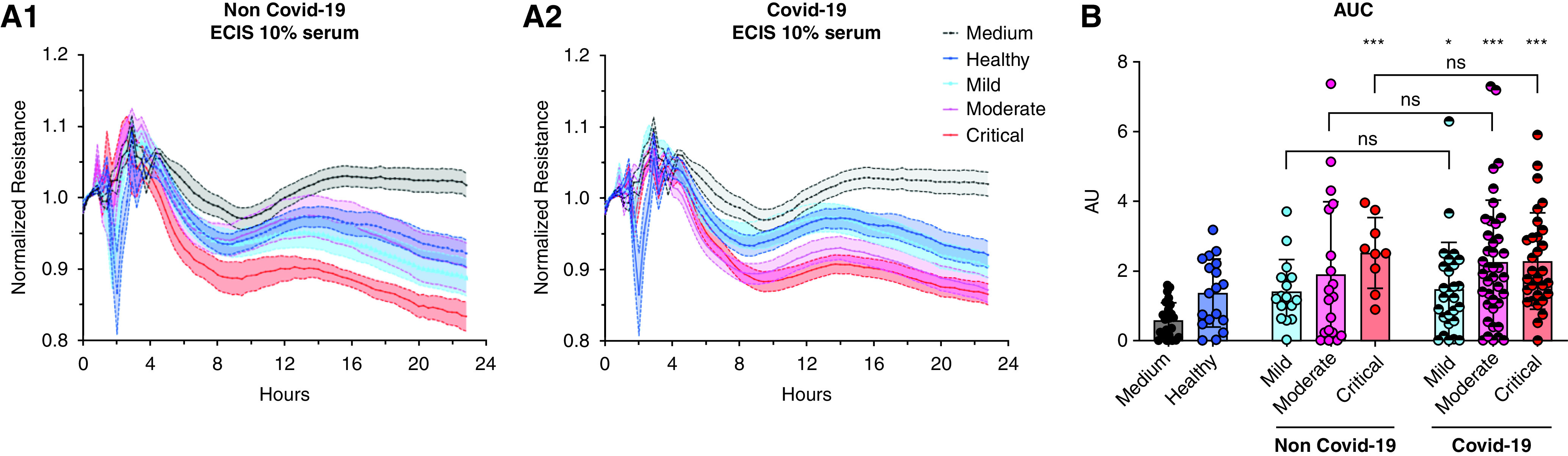
Rationale: Autopsy and biomarker studies suggest that endotheliopathy contributes to coronavirus disease (COVID-19)-associated acute respiratory distress syndrome. However, the effects of COVID-19 on the lung endothelium are not well defined. We hypothesized that the lung endotheliopathy of COVID-19 is caused by circulating host factors and direct endothelial infection by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Objectives: We aimed to determine the effects of SARS-CoV-2 or sera from patients with COVID-19 on the permeability and inflammatory activation of lung microvascular endothelial cells. Methods: Human lung microvascular endothelial cells were treated with live SARS-CoV-2; inactivated viral particles; or sera from patients with COVID-19, patients without COVID-19, and healthy volunteers. Permeability was determined by measuring transendothelial resistance to electrical current flow, where decreased resistance signifies increased permeability. Inflammatory mediators were quantified in culture supernatants. Endothelial biomarkers were quantified in patient sera. Measurements and Main Results: Viral PCR confirmed that SARS-CoV-2 enters and replicates in endothelial cells. Live SARS-CoV-2, but not dead virus or spike protein, induces endothelial permeability and secretion of plasminogen activator inhibitor 1 and vascular endothelial growth factor. There was substantial variability in the effects of SARS-CoV-2 on endothelial cells from different donors. Sera from patients with COVID-19 induced endothelial permeability, which correlated with disease severity. Serum levels of endothelial activation and injury biomarkers were increased in patients with COVID-19 and correlated with severity of illness. Conclusions: SARS-CoV-2 infects and dysregulates endothelial cell functions. Circulating factors in patients with COVID-19 also induce endothelial cell dysfunction. Our data point to roles for both systemic factors acting on lung endothelial cells and viral infection of endothelial cells in COVID-19-associated endotheliopathy.
Keywords: COVID-19; acute respiratory distress syndrome; endothelial permeability; lung endothelial injury.
Figures
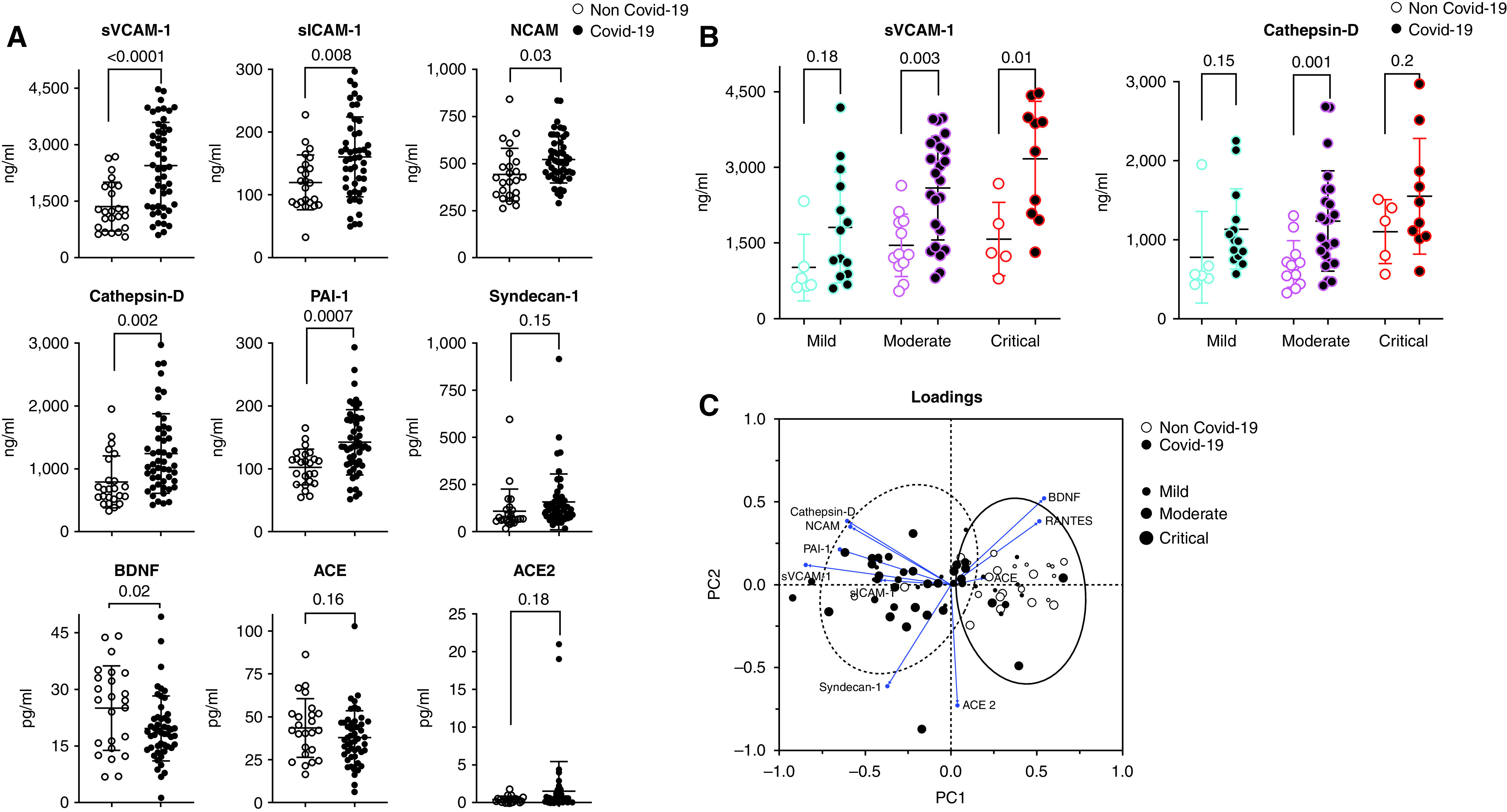
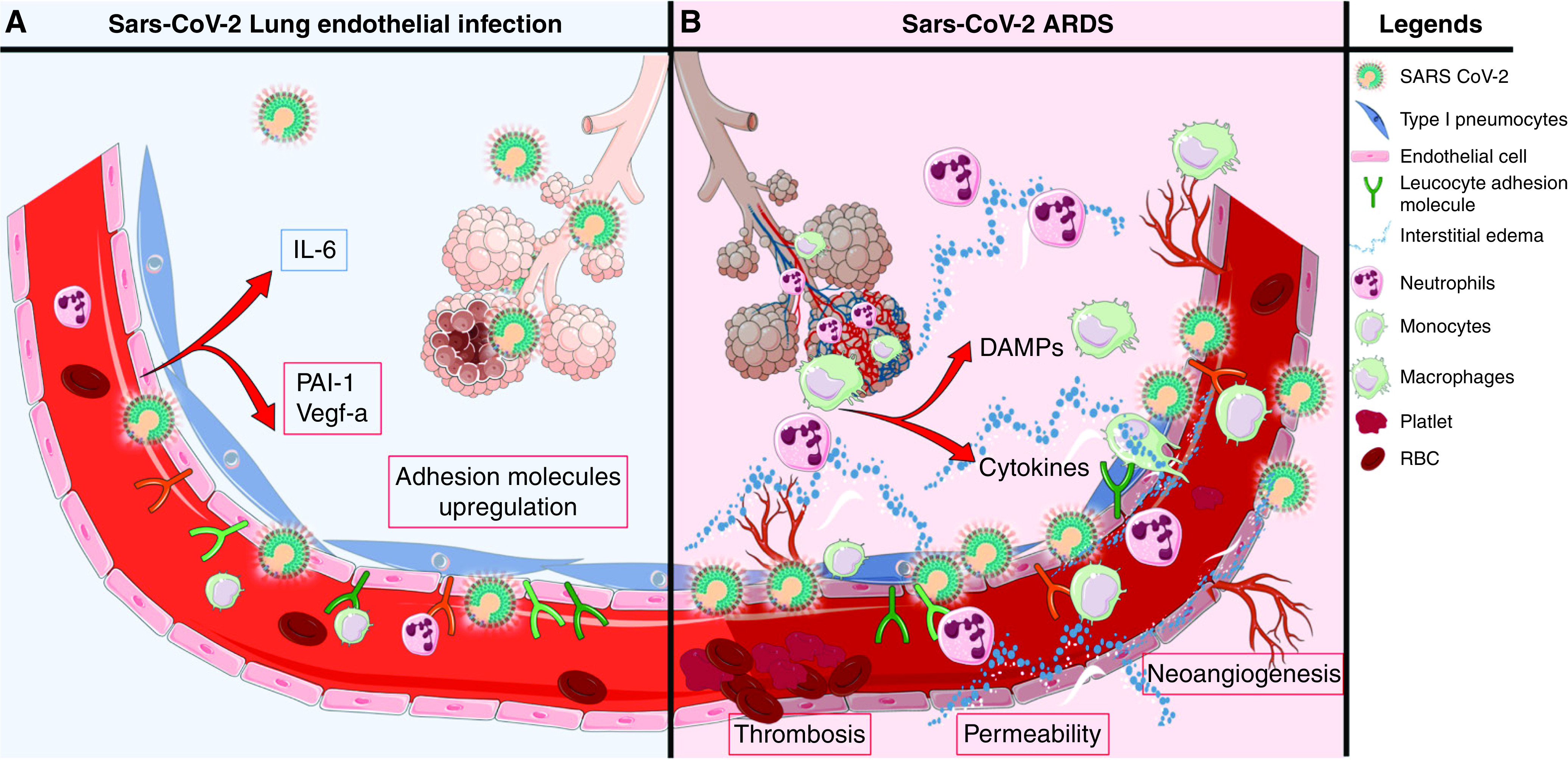
Comment in
-
Insights into Endotheliopathy in COVID-19.Am J Respir Crit Care Med. 2022 Oct 15;206(8):926-928. doi: 10.1164/rccm.202207-1258ED. Am J Respir Crit Care Med. 2022. PMID: 35819867 Free PMC article. No abstract available.
-
SARS-CoV2 Endotheliopathy: Insights from Single Cell RNAseq.Am J Respir Crit Care Med. 2022 Nov 1;206(9):1178-1179. doi: 10.1164/rccm.202206-1105LE. Am J Respir Crit Care Med. 2022. PMID: 35839476 Free PMC article. No abstract available.
Similar articles
-
SARS-CoV-2 Spike Proteins and Cell-Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis.Int J Mol Sci. 2022 Sep 9;23(18):10436. doi: 10.3390/ijms231810436. Int J Mol Sci. 2022. PMID: 36142345 Free PMC article.
-
Elevated Cytokine Levels in Plasma of Patients with SARS-CoV-2 Do Not Contribute to Pulmonary Microvascular Endothelial Permeability.Microbiol Spectr. 2022 Feb 23;10(1):e0167121. doi: 10.1128/spectrum.01671-21. Epub 2022 Feb 16. Microbiol Spectr. 2022. PMID: 35171047 Free PMC article.
-
The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells.Am J Physiol Lung Cell Mol Physiol. 2021 Aug 1;321(2):L477-L484. doi: 10.1152/ajplung.00223.2021. Epub 2021 Jun 22. Am J Physiol Lung Cell Mol Physiol. 2021. PMID: 34156871 Free PMC article.
-
Endothelial cell dysfunction, coagulation, and angiogenesis in coronavirus disease 2019 (COVID-19).Microvasc Res. 2021 Sep;137:104188. doi: 10.1016/j.mvr.2021.104188. Epub 2021 May 20. Microvasc Res. 2021. PMID: 34022205 Free PMC article. Review.
-
COVID-19 is a systemic vascular hemopathy: insight for mechanistic and clinical aspects.Angiogenesis. 2021 Nov;24(4):755-788. doi: 10.1007/s10456-021-09805-6. Epub 2021 Jun 28. Angiogenesis. 2021. PMID: 34184164 Free PMC article. Review.
Cited by
-
Levels of brain-derived neurotrophic factor (BDNF) among patients with COVID-19: a systematic review and meta-analysis.Eur Arch Psychiatry Clin Neurosci. 2024 Aug;274(5):1137-1152. doi: 10.1007/s00406-023-01681-z. Epub 2023 Aug 30. Eur Arch Psychiatry Clin Neurosci. 2024. PMID: 37646849
-
Longitudinal plasma proteomics reveals biomarkers of alveolar-capillary barrier disruption in critically ill COVID-19 patients.Nat Commun. 2024 Jan 25;15(1):744. doi: 10.1038/s41467-024-44986-w. Nat Commun. 2024. PMID: 38272877 Free PMC article.
-
Hydrogen Sulfide Ameliorates SARS-CoV-2-Associated Lung Endothelial Barrier Disruption.Biomedicines. 2023 Jun 22;11(7):1790. doi: 10.3390/biomedicines11071790. Biomedicines. 2023. PMID: 37509430 Free PMC article.
-
Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies.Acta Pharmacol Sin. 2023 Apr;44(4):695-709. doi: 10.1038/s41401-022-00998-0. Epub 2022 Oct 17. Acta Pharmacol Sin. 2023. PMID: 36253560 Free PMC article. Review.
-
Exploring extracellular vesicles as mediators of clinical disease and vehicles for viral therapeutics: Insights from the COVID-19 pandemic.Extracell Vesicles Circ Nucl Acids. 2022;3(3):172-188. doi: 10.20517/evcna.2022.19. Epub 2022 Jul 19. Extracell Vesicles Circ Nucl Acids. 2022. PMID: 35929616 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
