

Transcription feedback dynamics in the wake of cytoplasmic mRNA degradation shutdown
- PMID: 35640599
- PMCID: PMC9177992
- DOI: 10.1093/nar/gkac411
Transcription feedback dynamics in the wake of cytoplasmic mRNA degradation shutdown
Abstract
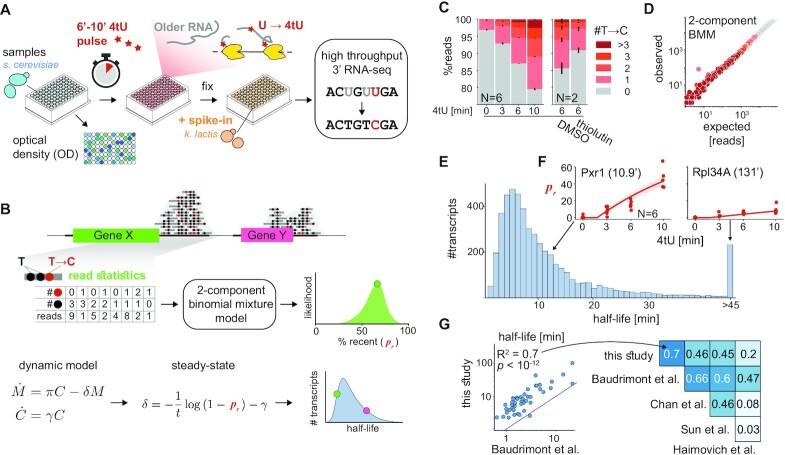
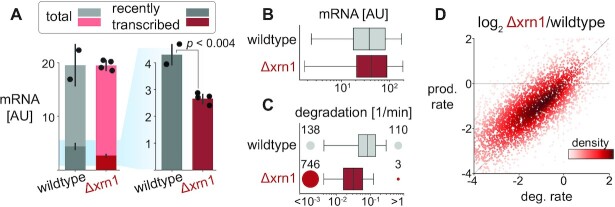
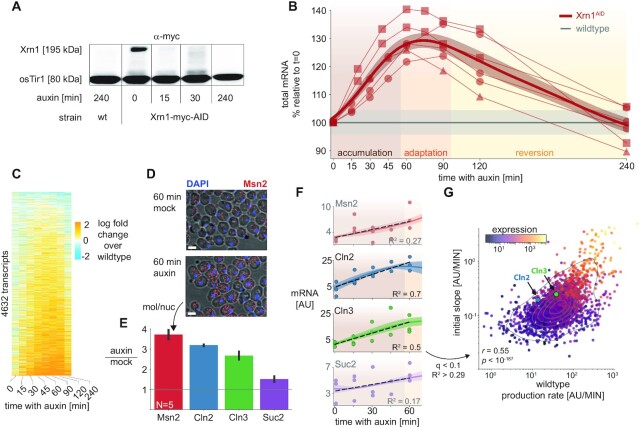
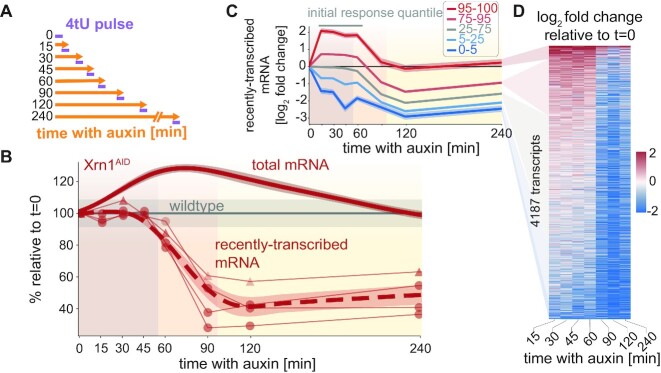
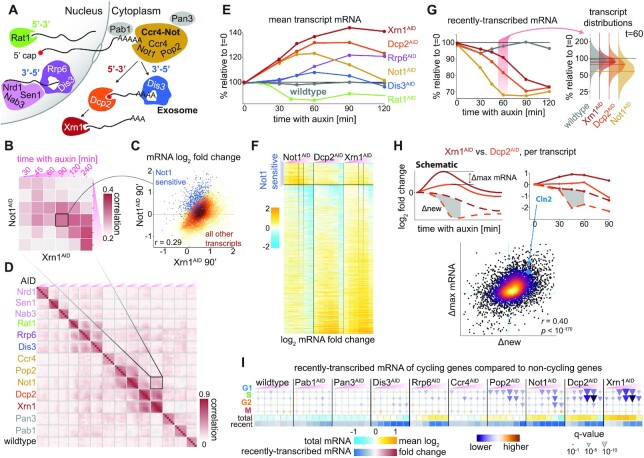
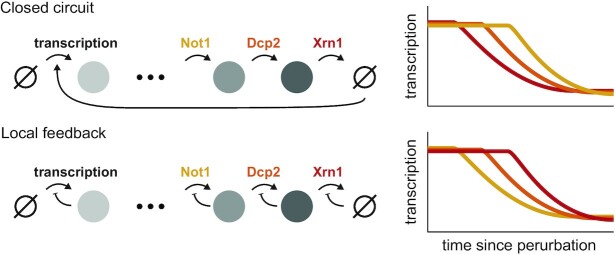
In the last decade, multiple studies demonstrated that cells maintain a balance of mRNA production and degradation, but the mechanisms by which cells implement this balance remain unknown. Here, we monitored cells' total and recently-transcribed mRNA profiles immediately following an acute depletion of Xrn1-the main 5'-3' mRNA exonuclease-which was previously implicated in balancing mRNA levels. We captured the detailed dynamics of the adaptation to rapid degradation of Xrn1 and observed a significant accumulation of mRNA, followed by a delayed global reduction in transcription and a gradual return to baseline mRNA levels. We found that this transcriptional response is not unique to Xrn1 depletion; rather, it is induced earlier when upstream factors in the 5'-3' degradation pathway are perturbed. Our data suggest that the mRNA feedback mechanism monitors the accumulation of inputs to the 5'-3' exonucleolytic pathway rather than its outputs.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
The yeast exoribonuclease Xrn1 and associated factors modulate RNA polymerase II processivity in 5' and 3' gene regions.J Biol Chem. 2020 Aug 14;295(33):11435-11454. doi: 10.1074/jbc.RA120.013426. Epub 2020 Jun 9. J Biol Chem. 2020. PMID: 32518159 Free PMC article.
-
Recruitment of Xrn1 to stress-induced genes allows efficient transcription by controlling RNA polymerase II backtracking.RNA Biol. 2021 Oct;18(10):1458-1474. doi: 10.1080/15476286.2020.1857521. Epub 2020 Dec 15. RNA Biol. 2021. PMID: 33258404 Free PMC article.
-
Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels.Mol Cell. 2013 Oct 10;52(1):52-62. doi: 10.1016/j.molcel.2013.09.010. Mol Cell. 2013. PMID: 24119399
-
Feedback to the central dogma: cytoplasmic mRNA decay and transcription are interdependent processes.Crit Rev Biochem Mol Biol. 2019 Aug;54(4):385-398. doi: 10.1080/10409238.2019.1679083. Epub 2019 Oct 27. Crit Rev Biochem Mol Biol. 2019. PMID: 31656086 Free PMC article. Review.
-
The roles of the exoribonucleases DIS3L2 and XRN1 in human disease.Biochem Soc Trans. 2016 Oct 15;44(5):1377-1384. doi: 10.1042/BST20160107. Biochem Soc Trans. 2016. PMID: 27911720 Review.
Cited by
-
The circular logic of mRNA homeostasis.Transcription. 2023 Nov;14(1-2):18-26. doi: 10.1080/21541264.2023.2183684. Epub 2023 Feb 26. Transcription. 2023. PMID: 36843061 Free PMC article. Review.
-
RNA-controlled nucleocytoplasmic shuttling of mRNA decay factors regulates mRNA synthesis and a novel mRNA decay pathway.Nat Commun. 2022 Nov 23;13(1):7184. doi: 10.1038/s41467-022-34417-z. Nat Commun. 2022. PMID: 36418294 Free PMC article.
-
Nuclear export is a limiting factor in eukaryotic mRNA metabolism.PLoS Comput Biol. 2024 May 16;20(5):e1012059. doi: 10.1371/journal.pcbi.1012059. eCollection 2024 May. PLoS Comput Biol. 2024. PMID: 38753883 Free PMC article.
-
Homeostasis of mRNA concentrations through coupling transcription, export, and degradation.iScience. 2024 Jul 18;27(8):110531. doi: 10.1016/j.isci.2024.110531. eCollection 2024 Aug 16. iScience. 2024. PMID: 39175768 Free PMC article.
-
Polysome propensity and tunable thresholds in coding sequence length enable differential mRNA stability.Sci Adv. 2023 Sep 29;9(39):eadh9545. doi: 10.1126/sciadv.adh9545. Epub 2023 Sep 27. Sci Adv. 2023. PMID: 37756413 Free PMC article.
References
-
- Hu W. The interplay between eukaryotic mRNA degradation and translation. Molecular cell biology. 2015; 1:346–353.
-
- Muhlrad D., Decker C.J., Parker R.. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′→3′ digestion of the transcript. Genes Dev. 1994; 8:855–866. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
