

Innate Immunity: A Balance between Disease and Adaption to Stress
- PMID: 35625664
- PMCID: PMC9138980
- DOI: 10.3390/biom12050737
Innate Immunity: A Balance between Disease and Adaption to Stress
Abstract
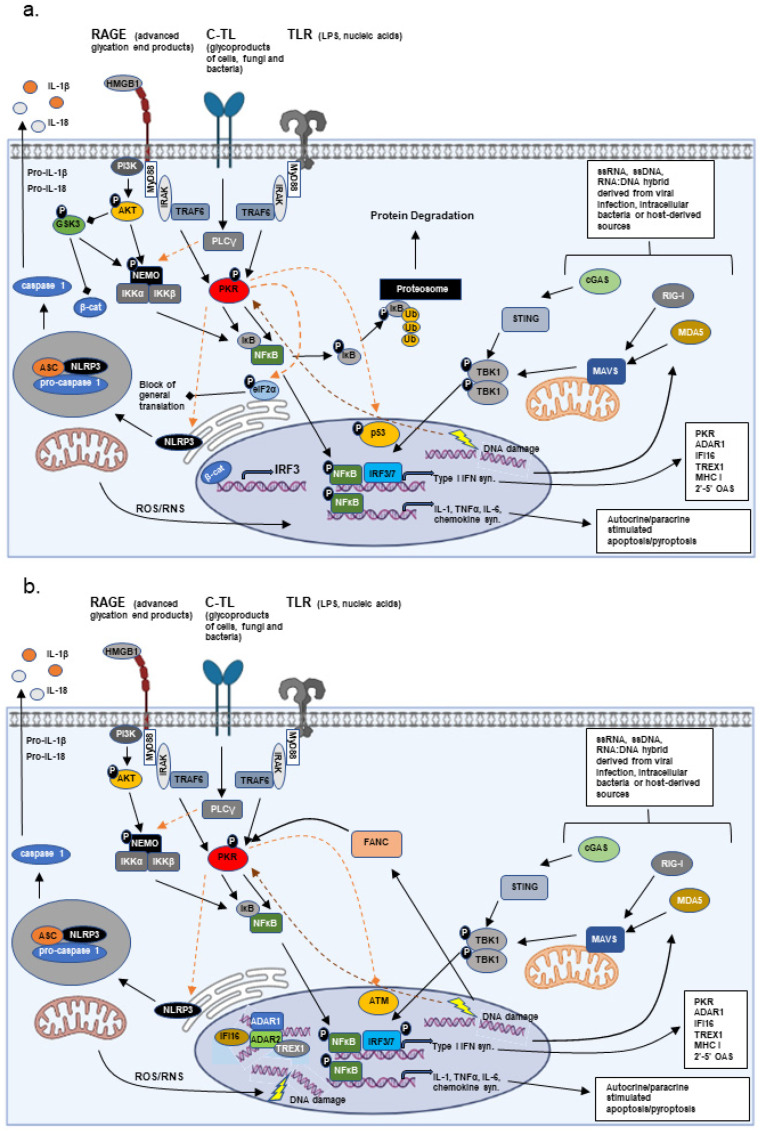
Since first being documented in ancient times, the relation of inflammation with injury and disease has evolved in complexity and causality. Early observations supported a cause (injury) and effect (inflammation) relationship, but the number of pathologies linked to chronic inflammation suggests that inflammation itself acts as a potent promoter of injury and disease. Additionally, results from studies over the last 25 years point to chronic inflammation and innate immune signaling as a critical link between stress (exogenous and endogenous) and adaptation. This brief review looks to highlight the role of the innate immune response in disease pathology, and recent findings indicating the innate immune response to chronic stresses as an influence in driving adaptation.
Keywords: Fanconi anemia; adaption; autoinflammation; cancer; inflammation; innate immunity; neuro-muscular degeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Innate Immune Nod1/RIP2 Signaling Is Essential for Cardiac Hypertrophy but Requires Mitochondrial Antiviral Signaling Protein for Signal Transductions and Energy Balance.Circulation. 2020 Dec 8;142(23):2240-2258. doi: 10.1161/CIRCULATIONAHA.119.041213. Epub 2020 Oct 19. Circulation. 2020. PMID: 33070627
-
The role of innate immune pathways in type 1 diabetes pathogenesis.Curr Opin Endocrinol Diabetes Obes. 2010 Apr;17(2):126-30. doi: 10.1097/MED.0b013e3283372819. Curr Opin Endocrinol Diabetes Obes. 2010. PMID: 20125005 Free PMC article. Review.
-
Nrf2 as regulator of innate immunity: A molecular Swiss army knife!Biotechnol Adv. 2018 Mar-Apr;36(2):358-370. doi: 10.1016/j.biotechadv.2017.12.012. Epub 2017 Dec 22. Biotechnol Adv. 2018. PMID: 29277308 Review.
-
Physiological roles of ASK family members in innate immunity and their involvement in pathogenesis of immune diseases.Adv Biol Regul. 2017 Dec;66:46-53. doi: 10.1016/j.jbior.2017.10.007. Epub 2017 Oct 19. Adv Biol Regul. 2017. PMID: 29122554 Review.
-
Identifying novel spatiotemporal regulators of innate immunity.Immunol Res. 2013 Mar;55(1-3):3-9. doi: 10.1007/s12026-012-8344-0. Immunol Res. 2013. PMID: 22926826 Free PMC article. Review.
Cited by
-
A Review on Asthma and Allergy: Current Understanding on Molecular Perspectives.J Clin Med. 2024 Sep 27;13(19):5775. doi: 10.3390/jcm13195775. J Clin Med. 2024. PMID: 39407835 Free PMC article. Review.
-
Helicase-like transcription factor (Hltf)-deletion activates Hmgb1-Rage axis and granzyme A-mediated killing of pancreatic β cells resulting in neonatal lethality.PLoS One. 2023 Aug 25;18(8):e0286109. doi: 10.1371/journal.pone.0286109. eCollection 2023. PLoS One. 2023. PMID: 37624843 Free PMC article.
-
Alternative Splicing, RNA Editing, and the Current Limits of Next Generation Sequencing.Genes (Basel). 2023 Jun 30;14(7):1386. doi: 10.3390/genes14071386. Genes (Basel). 2023. PMID: 37510291 Free PMC article.
References
-
- Granger D.N., Senchenkova E. Inflammation and the Microcirculation. Morgan & Claypool Life Sciences Publisher; San Rafael, CA, USA: 2010. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
