

Comparison of herpes simplex virus 1 genomic diversity between adult sexual transmission partners with genital infection
- PMID: 35587470
- PMCID: PMC9119503
- DOI: 10.1371/journal.ppat.1010437
Comparison of herpes simplex virus 1 genomic diversity between adult sexual transmission partners with genital infection
Abstract
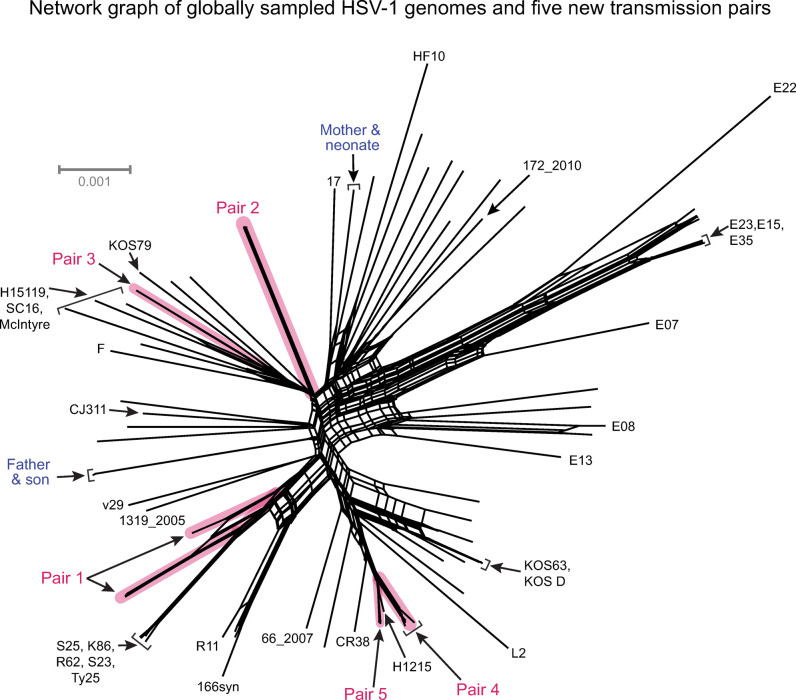
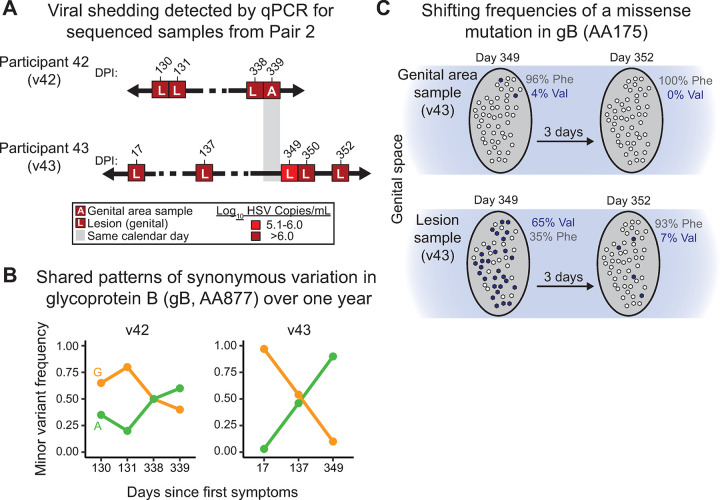
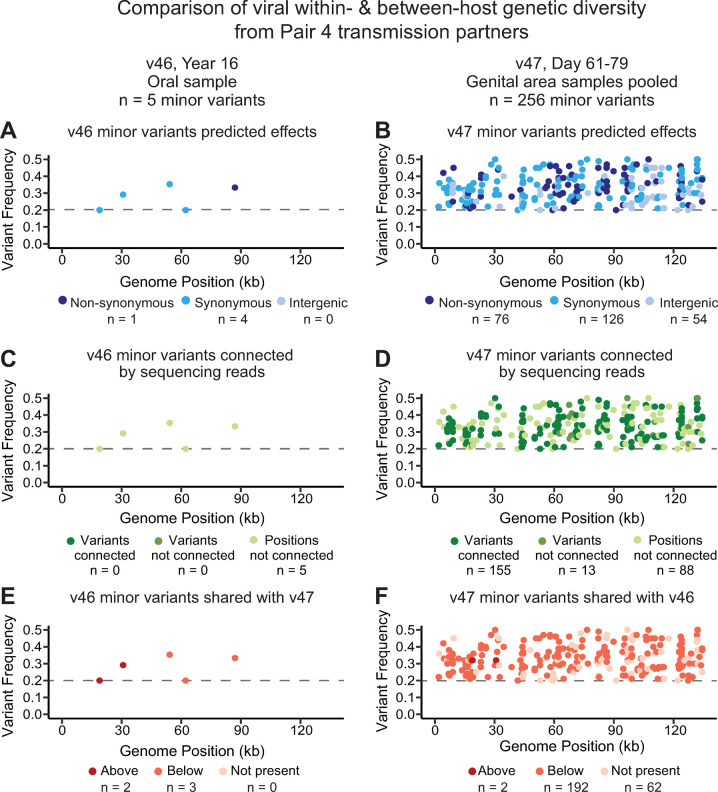
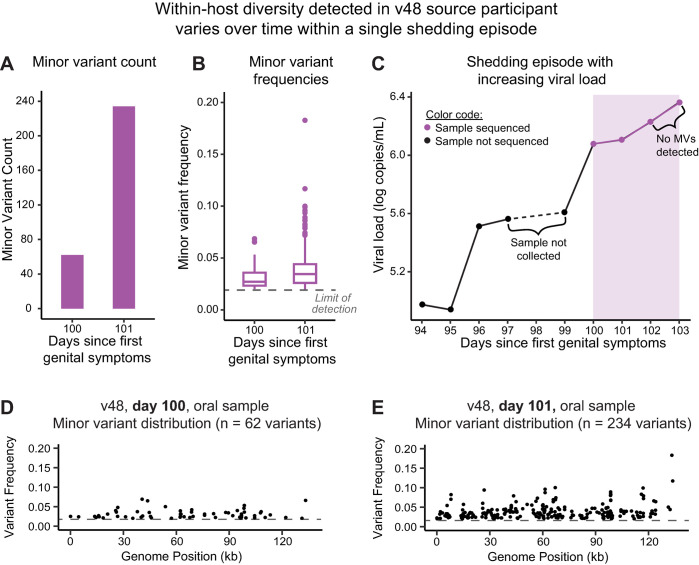
Herpes simplex virus (HSV) causes chronic infection in the human host, characterized by self-limited episodes of mucosal shedding and lesional disease, with latent infection of neuronal ganglia. The epidemiology of genital herpes has undergone a significant transformation over the past two decades, with the emergence of HSV-1 as a leading cause of first-episode genital herpes in many countries. Though dsDNA viruses are not expected to mutate quickly, it is not yet known to what degree the HSV-1 viral population in a natural host adapts over time, or how often viral population variants are transmitted between hosts. This study provides a comparative genomics analysis for 33 temporally-sampled oral and genital HSV-1 genomes derived from five adult sexual transmission pairs. We found that transmission pairs harbored consensus-level viral genomes with near-complete conservation of nucleotide identity. Examination of within-host minor variants in the viral population revealed both shared and unique patterns of genetic diversity between partners, and between anatomical niches. Additionally, genetic drift was detected from spatiotemporally separated samples in as little as three days. These data expand our prior understanding of the complex interaction between HSV-1 genomics and population dynamics after transmission to new infected persons.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: CJ reports funding from UpToDate (royalties), AbbVie (consulting), Gilead (consulting), MedPace (DSMB), NIH and CDC. AW reports funding from NIH, Sanofi (grants), Aicuris (consulting), X-vax (consulting), Auritec (consulting), GSK (grants), Merck (DSMB), Crozet (consulting), VIR (consulting), and UptoDate (royalties).
Figures




Similar articles
-
Viral Shedding 1 Year Following First-Episode Genital HSV-1 Infection.JAMA. 2022 Nov 1;328(17):1730-1739. doi: 10.1001/jama.2022.19061. JAMA. 2022. PMID: 36272098 Free PMC article.
-
Genotypic and Phenotypic Diversity of Herpes Simplex Virus 2 within the Infected Neonatal Population.mSphere. 2019 Feb 27;4(1):e00590-18. doi: 10.1128/mSphere.00590-18. mSphere. 2019. PMID: 30814317 Free PMC article.
-
A holistic perspective on herpes simplex virus (HSV) ecology and evolution.Adv Virus Res. 2021;110:27-57. doi: 10.1016/bs.aivir.2021.05.001. Epub 2021 Jun 26. Adv Virus Res. 2021. PMID: 34353481 Free PMC article. Review.
-
Molecular Evolution of Herpes Simplex Virus 2 Complete Genomes: Comparison between Primary and Recurrent Infections.J Virol. 2017 Nov 14;91(23):e00942-17. doi: 10.1128/JVI.00942-17. Print 2017 Dec 1. J Virol. 2017. PMID: 28931680 Free PMC article.
-
Genital Herpes Infection: Progress and Problems.Infect Dis Clin North Am. 2023 Jun;37(2):351-367. doi: 10.1016/j.idc.2023.02.011. Infect Dis Clin North Am. 2023. PMID: 37105647 Review.
Cited by
-
Multi-phenotype analysis for enhanced classification of 11 herpes simplex virus 1 strains.J Gen Virol. 2022 Oct;103(10):001780. doi: 10.1099/jgv.0.001780. J Gen Virol. 2022. PMID: 36264606 Free PMC article.
-
Evolutionary Dynamics of Accelerated Antiviral Resistance Development in Hypermutator Herpesvirus.Mol Biol Evol. 2024 Jul 3;41(7):msae119. doi: 10.1093/molbev/msae119. Mol Biol Evol. 2024. PMID: 38879872 Free PMC article.
-
Phylogenetic and Genomic Characterization of Whole Genome Sequences of Ocular Herpes Simplex Virus Type 1 Isolates Identifies Possible Virulence Determinants in Humans.Invest Ophthalmol Vis Sci. 2023 Jul 3;64(10):16. doi: 10.1167/iovs.64.10.16. Invest Ophthalmol Vis Sci. 2023. PMID: 37450309 Free PMC article.
-
Synonymous nucleotide changes drive papillomavirus evolution.Tumour Virus Res. 2022 Dec;14:200248. doi: 10.1016/j.tvr.2022.200248. Epub 2022 Oct 17. Tumour Virus Res. 2022. PMID: 36265836 Free PMC article. Review.
-
Viral Shedding 1 Year Following First-Episode Genital HSV-1 Infection.JAMA. 2022 Nov 1;328(17):1730-1739. doi: 10.1001/jama.2022.19061. JAMA. 2022. PMID: 36272098 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
