

Multifaceted regulation and functions of 53BP1 in NHEJ‑mediated DSB repair (Review)
- PMID: 35583003
- PMCID: PMC9162042
- DOI: 10.3892/ijmm.2022.5145
Multifaceted regulation and functions of 53BP1 in NHEJ‑mediated DSB repair (Review)
Abstract
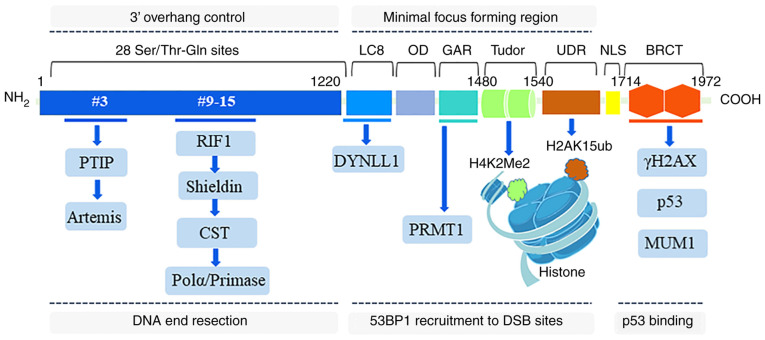
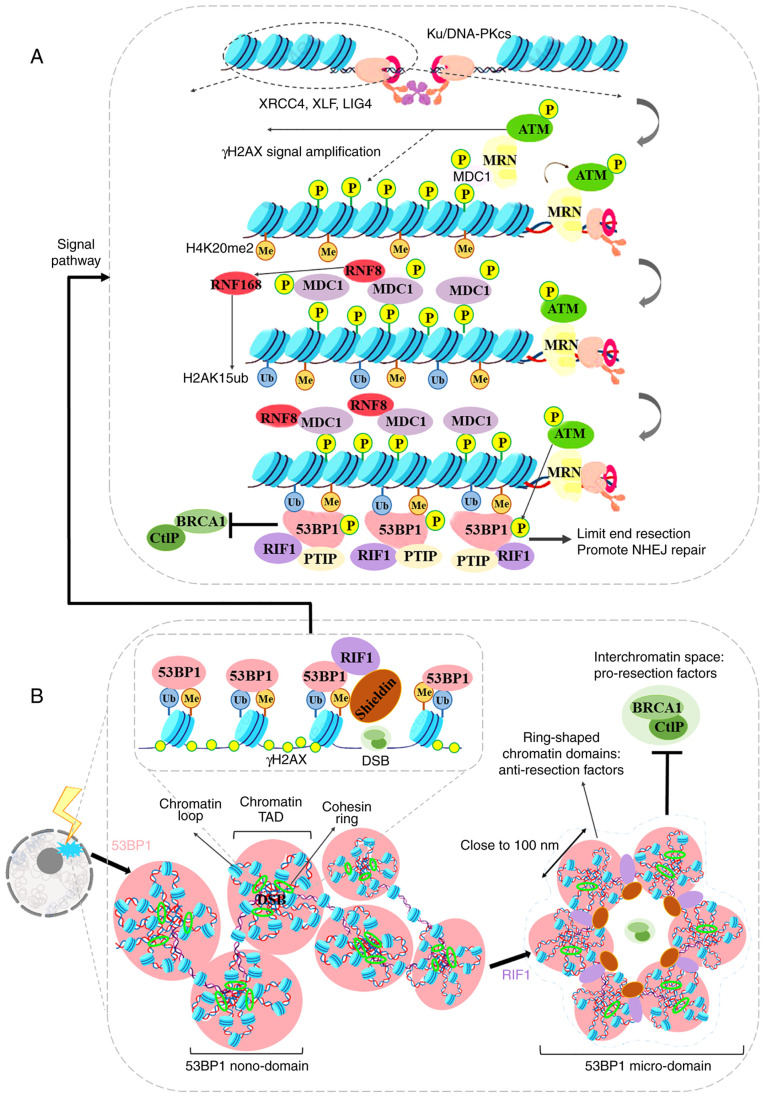
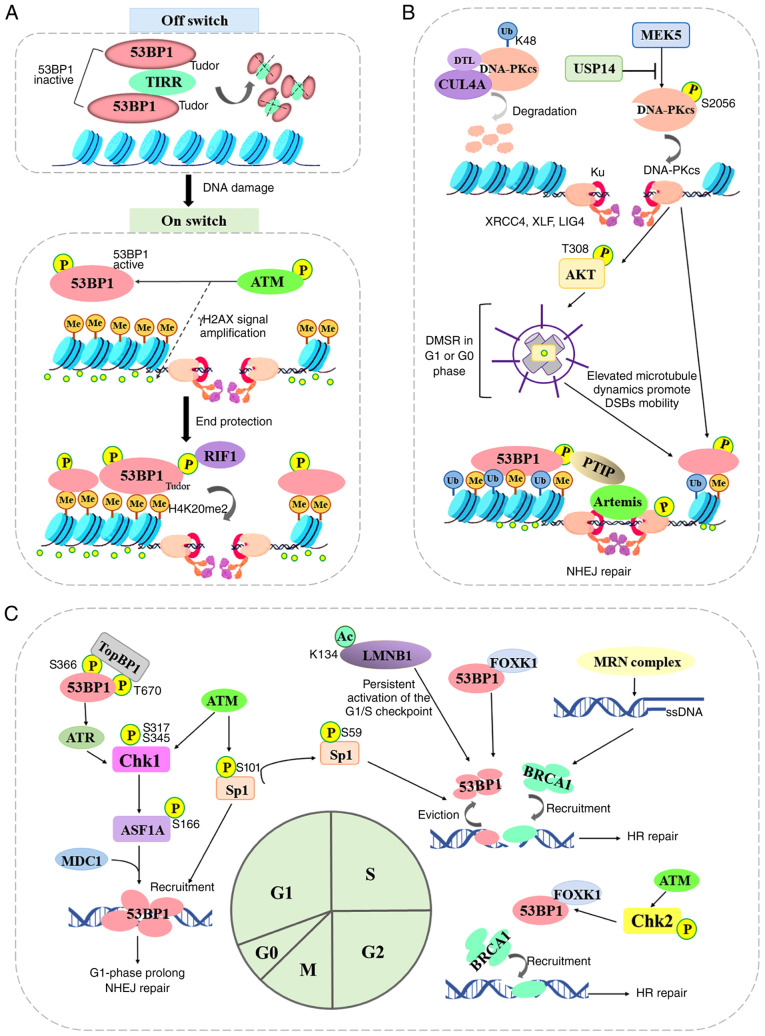
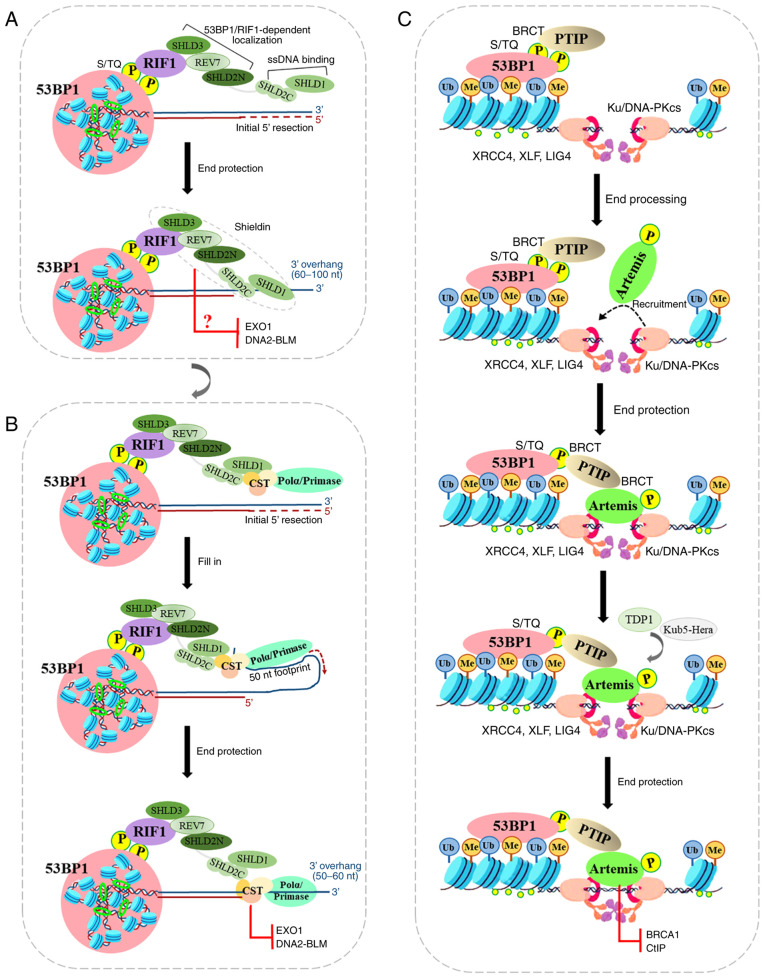
The repair of DNA double‑strand breaks (DSBs) is crucial for the preservation of genomic integrity and the maintenance of cellular homeostasis. Non‑homologous DNA end joining (NHEJ) is the predominant repair mechanism for any type of DNA DSB during the majority of the cell cycle. NHEJ defects regulate tumor sensitivity to ionizing radiation and anti‑neoplastic agents, resulting in immunodeficiencies and developmental abnormalities in malignant cells. p53‑binding protein 1 (53BP1) is a key mediator involved in DSB repair, which functions to maintain a balance in the repair pathway choices and in preserving genomic stability. 53BP1 promotes DSB repair via NHEJ and antagonizes DNA end overhang resection. At present, novel lines of evidence have revealed the molecular mechanisms underlying the recruitment of 53BP1 and DNA break‑responsive effectors to DSB sites, and the promotion of NHEJ‑mediated DSB repair via 53BP1, while preventing homologous recombination. In the present review article, recent advances made in the elucidation of the structural and functional characteristics of 53BP1, the mechanisms of 53BP1 recruitment and interaction with the reshaping of the chromatin architecture around DSB sites, the post‑transcriptional modifications of 53BP1, and the up‑ and downstream pathways of 53BP1 are discussed. The present review article also focuses on the application perspectives, current challenges and future directions of 53BP1 research.
Keywords: DNA double strand break; Pax transactivation domain‑interacting protein; RAP1‑interacting factor 1; non‑homologous end joining; p53‑binding protein 1.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Roles for 53BP1 in the repair of radiation-induced DNA double strand breaks.DNA Repair (Amst). 2020 Sep;93:102915. doi: 10.1016/j.dnarep.2020.102915. DNA Repair (Amst). 2020. PMID: 33087281 Review.
-
Regulation of repair pathway choice at two-ended DNA double-strand breaks.Mutat Res. 2017 Oct;803-805:51-55. doi: 10.1016/j.mrfmmm.2017.07.011. Epub 2017 Jul 29. Mutat Res. 2017. PMID: 28781144 Review.
-
53BP1: Keeping It under Control, Even at a Distance from DNA Damage.Genes (Basel). 2022 Dec 16;13(12):2390. doi: 10.3390/genes13122390. Genes (Basel). 2022. PMID: 36553657 Free PMC article. Review.
-
Regulation of DNA double-strand break repair pathway choice: a new focus on 53BP1.J Zhejiang Univ Sci B. 2021 Jan 15;22(1):38-46. doi: 10.1631/jzus.B2000306. J Zhejiang Univ Sci B. 2021. PMID: 33448186 Free PMC article. Review.
-
Roles for the DNA-PK complex and 53BP1 in protecting ends from resection during DNA double-strand break repair.J Radiat Res. 2020 Sep 8;61(5):718-726. doi: 10.1093/jrr/rraa053. J Radiat Res. 2020. PMID: 32779701 Free PMC article. Review.
Cited by
-
Fance deficiency impaired DNA damage repair of prospermatogonia and altered the repair dynamics of spermatocytes.Reprod Biol Endocrinol. 2024 Aug 29;22(1):113. doi: 10.1186/s12958-024-01284-w. Reprod Biol Endocrinol. 2024. PMID: 39210375 Free PMC article.
-
Ubiquitination Links DNA Damage and Repair Signaling to Cancer Metabolism.Int J Mol Sci. 2023 May 8;24(9):8441. doi: 10.3390/ijms24098441. Int J Mol Sci. 2023. PMID: 37176148 Free PMC article. Review.
-
Discovery of a 53BP1 Small Molecule Antagonist Using a Focused DNA-Encoded Library Screen.J Med Chem. 2023 Oct 26;66(20):14133-14149. doi: 10.1021/acs.jmedchem.3c01192. Epub 2023 Oct 2. J Med Chem. 2023. PMID: 37782247 Free PMC article.
-
NEAT1 modulates the TIRR/53BP1 complex to maintain genome integrity.Nat Commun. 2024 Sep 30;15(1):8438. doi: 10.1038/s41467-024-52862-w. Nat Commun. 2024. PMID: 39349456 Free PMC article.
-
DNA Damage Repair and Current Therapeutic Approaches in Gastric Cancer: A Comprehensive Review.Front Genet. 2022 Aug 12;13:931866. doi: 10.3389/fgene.2022.931866. eCollection 2022. Front Genet. 2022. PMID: 36035159 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
