

Emerging actionable targets to treat therapy-resistant colorectal cancers
- PMID: 35582524
- PMCID: PMC8992594
- DOI: 10.20517/cdr.2021.96
Emerging actionable targets to treat therapy-resistant colorectal cancers
Abstract
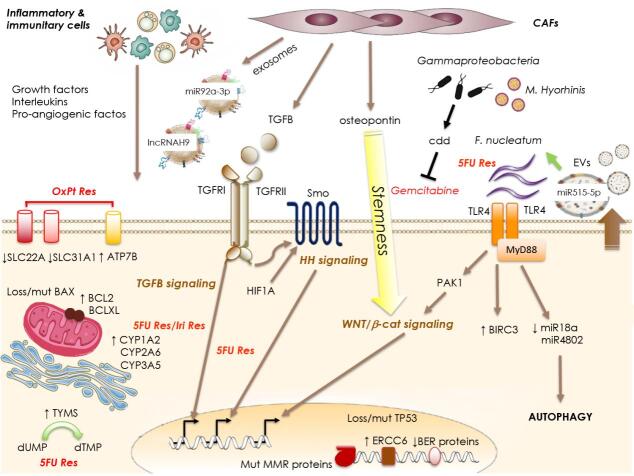
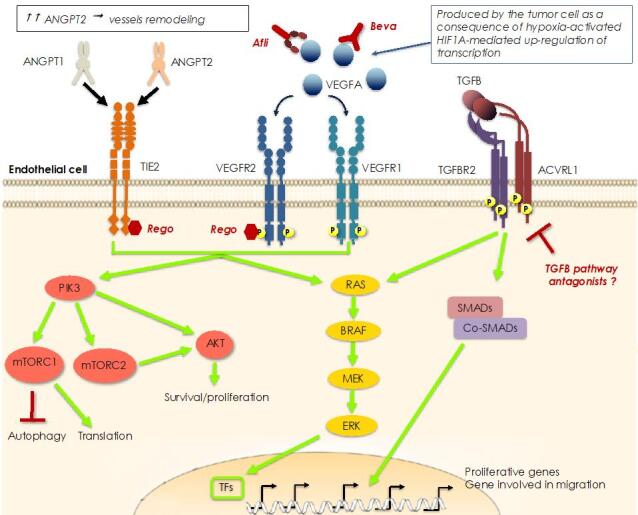
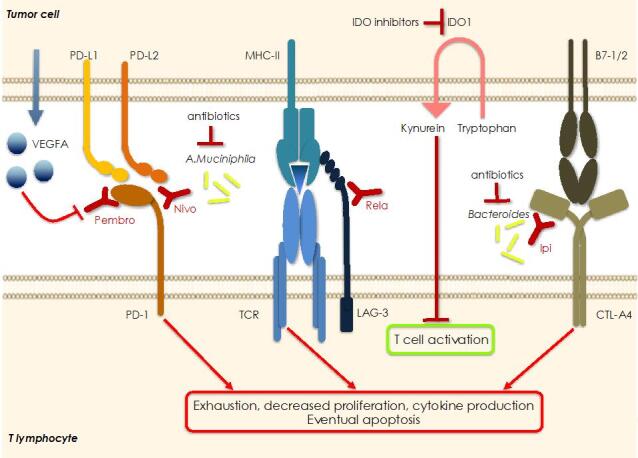
In the last two decades major improvements have been reached in the early diagnosis of colorectal cancer (CRC) and, besides chemotherapy, an ampler choice of therapeutic approaches is now available, including targeted and immunotherapy. Despite that, CRC remains a "big killer" mainly due to the development of resistance to therapies, especially when the disease is diagnosed after it is already metastatic. At the same time, our knowledge of the mechanisms underlying resistance has been rapidly expanding which allows the development of novel therapeutic options in order to overcome it. As far as resistance to chemotherapy is concerned, several contributors have been identified such as: intake/efflux systems upregulation; alterations in the DNA damage response, due to defect in the DNA checkpoint and repair systems; dysregulation of the expression of apoptotic/anti-apoptotic members of the BCL2 family; overexpression of oncogenic kinases; the presence of cancer stem cells; and the composition of the tumoral microenvironment and that of the gut microbiota. Interestingly, several mechanisms are also involved in the resistance to targeted and/or immunotherapy. For example, overexpression and/or hyperactivation and/or amplification of oncogenic kinases can sustain resistance to targeted therapy whereas the composition of the gut microbiota, as well as that of the tumoral niche, and defects in DNA repair systems are crucial for determining the response to immunotherapy. In this review we will make an overview of the main resistance mechanisms identified so far and of the new therapeutic approaches to overcome it.
Keywords: BRAF; Colorectal cancer; EGFR; ERBB2; MET; chemotherapy; gut microbiota; immune checkpoint inhibitors; kinase inhibitors; resistance; target therapy.
© The Author(s) 2022.
Conflict of interest statement
Both authors declared that there are no conflicts of interest.
Figures

Similar articles
-
Therapeutic Strategies in Diseases of the Digestive Tract - 2015 and Beyond Targeted Therapies in Colon Cancer Today and Tomorrow.Dig Dis. 2016;34(5):574-9. doi: 10.1159/000445267. Epub 2016 Jun 22. Dig Dis. 2016. PMID: 27332557
-
Resistance to anti-EGFR therapies in metastatic colorectal cancer: underlying mechanisms and reversal strategies.J Exp Clin Cancer Res. 2021 Oct 18;40(1):328. doi: 10.1186/s13046-021-02130-2. J Exp Clin Cancer Res. 2021. PMID: 34663410 Free PMC article. Review.
-
Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation-A Special Reference to Colorectal Cancer.Cancers (Basel). 2020 Dec 25;13(1):43. doi: 10.3390/cancers13010043. Cancers (Basel). 2020. PMID: 33375686 Free PMC article. Review.
-
Future perspectives in melanoma research: meeting report from the "Melanoma Bridge": Napoli, December 3rd-6th 2014.J Transl Med. 2015 Nov 30;13:374. doi: 10.1186/s12967-015-0736-1. J Transl Med. 2015. PMID: 26619946 Free PMC article.
-
Overview of resistance to systemic therapy in patients with breast cancer.Adv Exp Med Biol. 2007;608:1-22. doi: 10.1007/978-0-387-74039-3_1. Adv Exp Med Biol. 2007. PMID: 17993229 Review.
Cited by
-
Circular RNA circNCOA3 promotes tumor progression and anti-PD-1 resistance in colorectal cancer.Cancer Drug Resist. 2024 Mar 13;7:9. doi: 10.20517/cdr.2023.151. eCollection 2024. Cancer Drug Resist. 2024. PMID: 38510750 Free PMC article.
-
Gastrointestinal Microbiota and Breast Cancer Chemotherapy Interactions: A Systematic Review.Cureus. 2022 Nov 18;14(11):e31648. doi: 10.7759/cureus.31648. eCollection 2022 Nov. Cureus. 2022. PMID: 36540440 Free PMC article. Review.
-
AEG-1 as a Novel Therapeutic Target in Colon Cancer: A Study from Silencing AEG-1 in BALB/c Mice to Large Data Analysis.Curr Gene Ther. 2024;24(4):307-320. doi: 10.2174/0115665232273077240104045022. Curr Gene Ther. 2024. PMID: 38783530
-
TGM2, HMGA2, FXYD3, and LGALS4 genes as biomarkers in acquired oxaliplatin resistance of human colorectal cancer: A systems biology approach.PLoS One. 2023 Aug 3;18(8):e0289535. doi: 10.1371/journal.pone.0289535. eCollection 2023. PLoS One. 2023. PMID: 37535601 Free PMC article.
-
Insights into fourth generation selective inhibitors of (C797S) EGFR mutation combating non-small cell lung cancer resistance: a critical review.RSC Adv. 2023 Jun 21;13(27):18825-18853. doi: 10.1039/d3ra02347h. eCollection 2023 Jun 15. RSC Adv. 2023. PMID: 37350862 Free PMC article. Review.
References
-
- Colorectal cancer: statistics. Available from: https://www.cancer.net/cancer-types/colorectal-cancer/statistics. [Last accessed on 23 Dec 2021]
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous