

'Fly-ing' from rare to common neurodegenerative disease mechanisms
- PMID: 35484057
- PMCID: PMC9378361
- DOI: 10.1016/j.tig.2022.03.018
'Fly-ing' from rare to common neurodegenerative disease mechanisms
Abstract
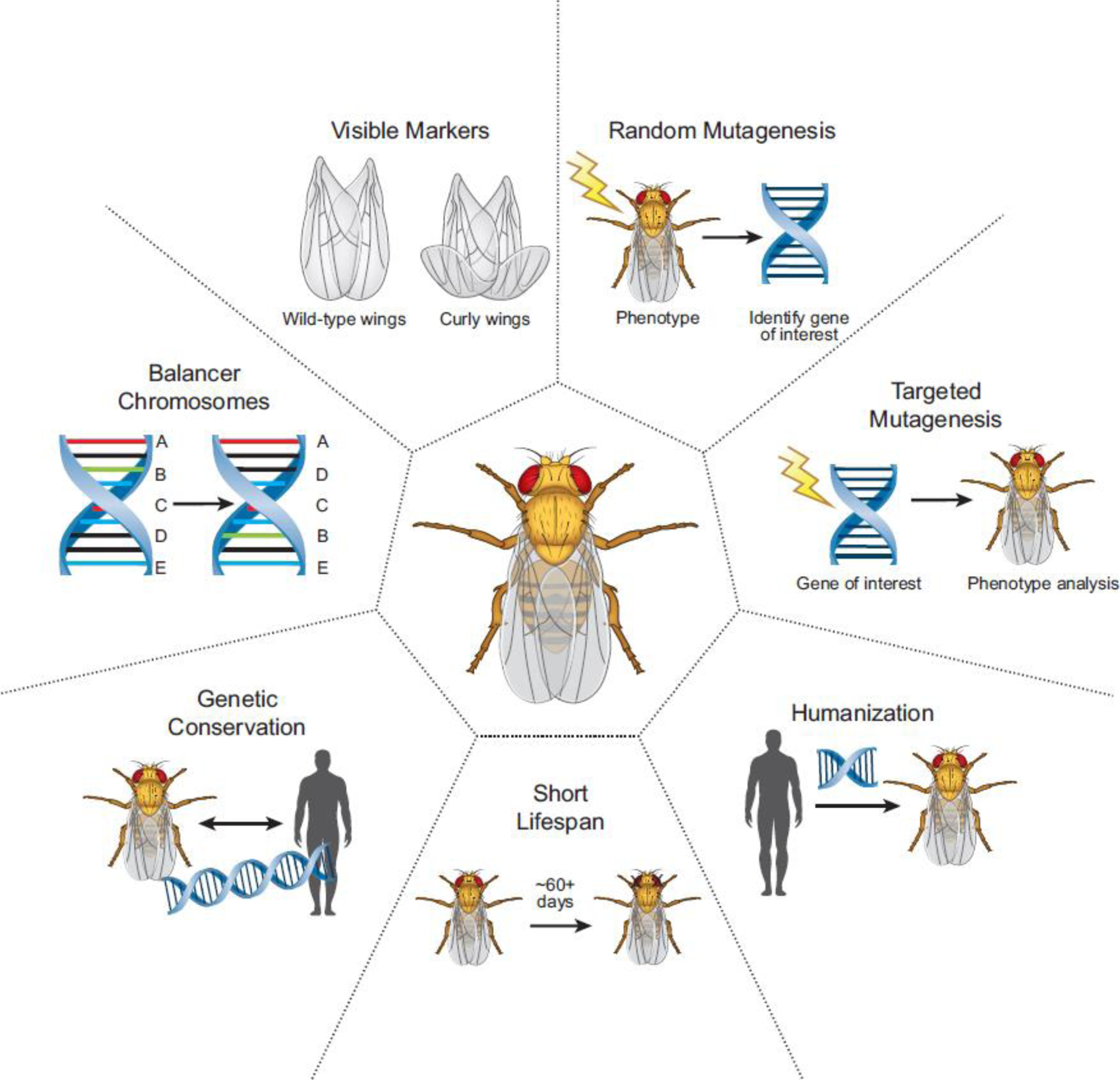
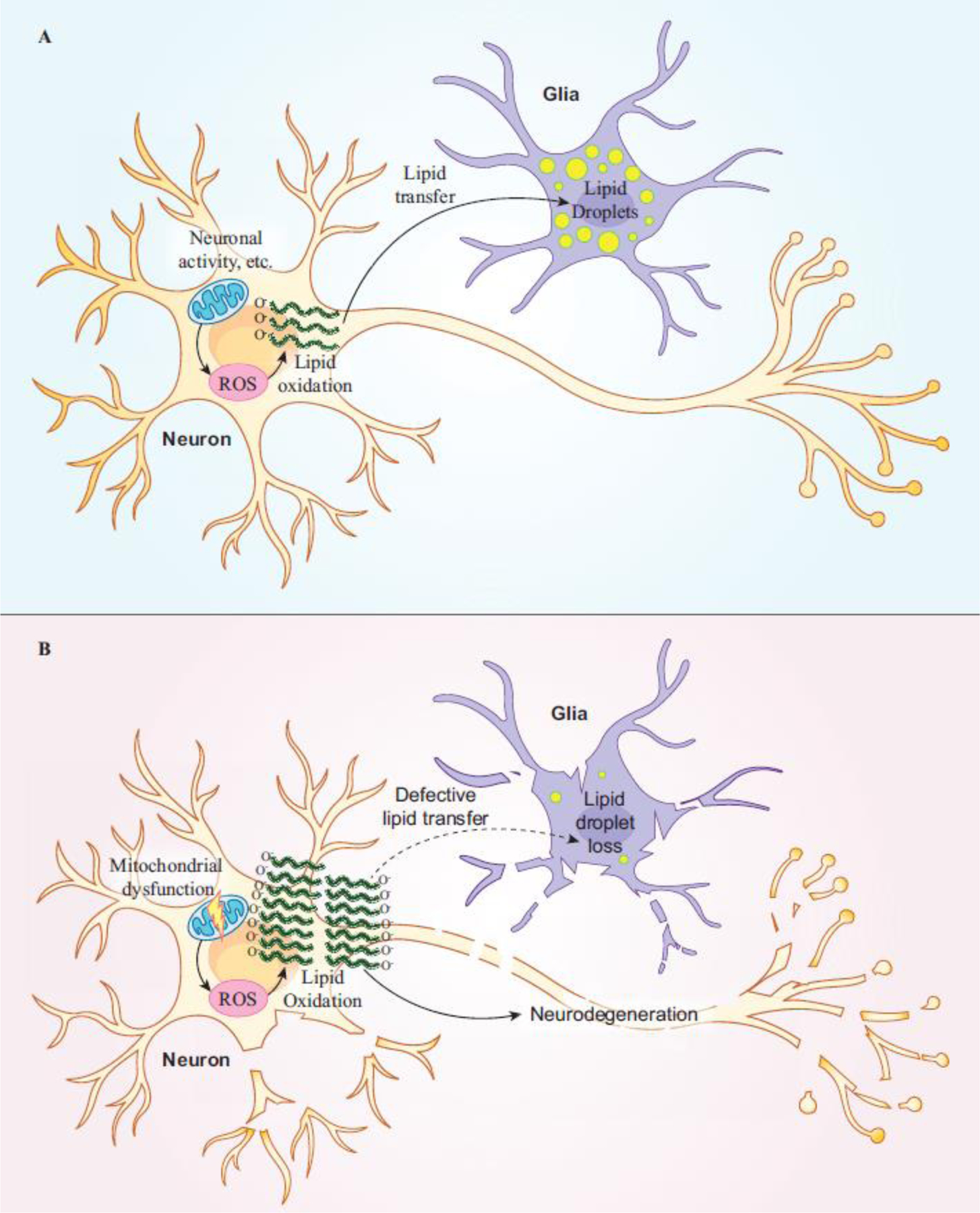
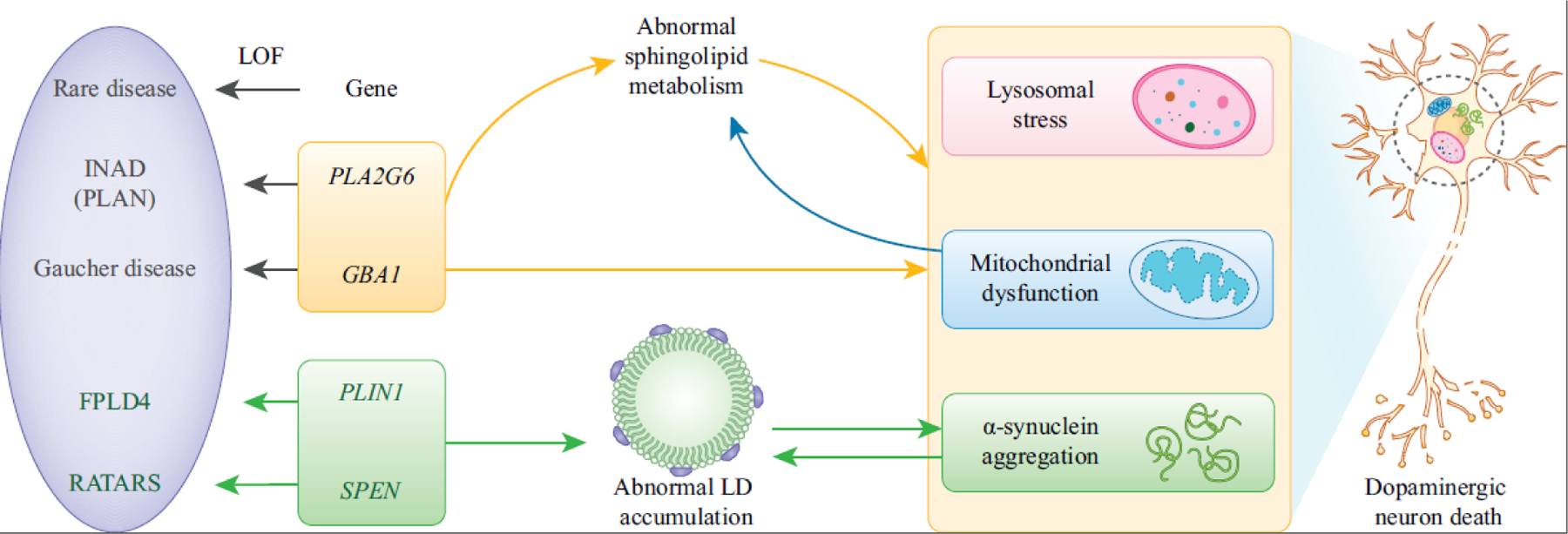
Advances in genome sequencing have enabled researchers and clinicians to probe vast numbers of human variants to distinguish pathogenic from benign variants. Model organisms have been crucial in variant assessment and in delineating the molecular mechanisms of some of the diseases caused by these variants. The fruit fly, Drosophila melanogaster, has played a valuable role in this endeavor, taking advantage of its genetic technologies and established biological knowledge. We highlight the utility of the fly in studying the function of genes associated with rare neurological diseases that have led to a better understanding of common disease mechanisms. We emphasize that shared themes emerge among disease mechanisms, including the importance of lipids, in two prominent neurodegenerative diseases: Alzheimer's disease (AD) and Parkinson's disease (PD).
Keywords: Alzheimer's disease; Drosophila; Parkinson's disease; lipid; mitochondria; neurodegeneration.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no conflicts of interest.
Figures
Similar articles
-
Drosophila as a model for human neurodegenerative disease.Annu Rev Genet. 2005;39:153-71. doi: 10.1146/annurev.genet.39.110304.095804. Annu Rev Genet. 2005. PMID: 16285856 Review.
-
Drosophila models of neurodegenerative disease.NeuroRx. 2005 Jul;2(3):438-46. doi: 10.1602/neurorx.2.3.438. NeuroRx. 2005. PMID: 16389307 Free PMC article. Review.
-
Drosophila melanogaster in the study of human neurodegeneration.CNS Neurol Disord Drug Targets. 2010 Aug;9(4):504-23. doi: 10.2174/187152710791556104. CNS Neurol Disord Drug Targets. 2010. PMID: 20522007 Free PMC article. Review.
-
An evaluation of Drosophila as a model system for studying tauopathies such as Alzheimer's disease.J Neurosci Methods. 2019 May 1;319:77-88. doi: 10.1016/j.jneumeth.2019.01.001. Epub 2019 Jan 8. J Neurosci Methods. 2019. PMID: 30633936
-
Identifying the Association Between Alzheimer's Disease and Parkinson's Disease Using Genome-Wide Association Studies and Protein-Protein Interaction Network.Mol Neurobiol. 2015 Dec;52(3):1629-1636. doi: 10.1007/s12035-014-8946-8. Epub 2014 Nov 5. Mol Neurobiol. 2015. PMID: 25370933
Cited by
-
Unraveling the link between neuropathy target esterase NTE/SWS, lysosomal storage diseases, inflammation, abnormal fatty acid metabolism, and leaky brain barrier.Elife. 2024 Apr 25;13:e98020. doi: 10.7554/eLife.98020. Elife. 2024. PMID: 38660940 Free PMC article.
-
An optimized temporally controlled Gal4 system in Drosophila reveals degeneration caused by adult-onset neuronal Vps13D knockdown.Front Neurosci. 2023 Jun 29;17:1204068. doi: 10.3389/fnins.2023.1204068. eCollection 2023. Front Neurosci. 2023. PMID: 37457002 Free PMC article.
-
Advanced Cellular Models for Rare Disease Study: Exploring Neural, Muscle and Skeletal Organoids.Int J Mol Sci. 2024 Jan 13;25(2):1014. doi: 10.3390/ijms25021014. Int J Mol Sci. 2024. PMID: 38256087 Free PMC article. Review.
-
Neuronal LRP4 directs the development, maturation, and cytoskeletal organization of peripheral synapses.bioRxiv [Preprint]. 2023 Nov 8:2023.11.03.564481. doi: 10.1101/2023.11.03.564481. bioRxiv. 2023. Update in: Development. 2024 Jun 1;151(11):dev202517. doi: 10.1242/dev.202517 PMID: 37961323 Free PMC article. Updated. Preprint.
-
Genetic modifiers of synucleinopathies-lessons from experimental models.Oxf Open Neurosci. 2023 Mar 9;2:kvad001. doi: 10.1093/oons/kvad001. eCollection 2023. Oxf Open Neurosci. 2023. PMID: 38596238 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
