

The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development
- PMID: 35473814
- PMCID: PMC9464682
- DOI: 10.1016/j.tcb.2022.03.012
The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development
Abstract
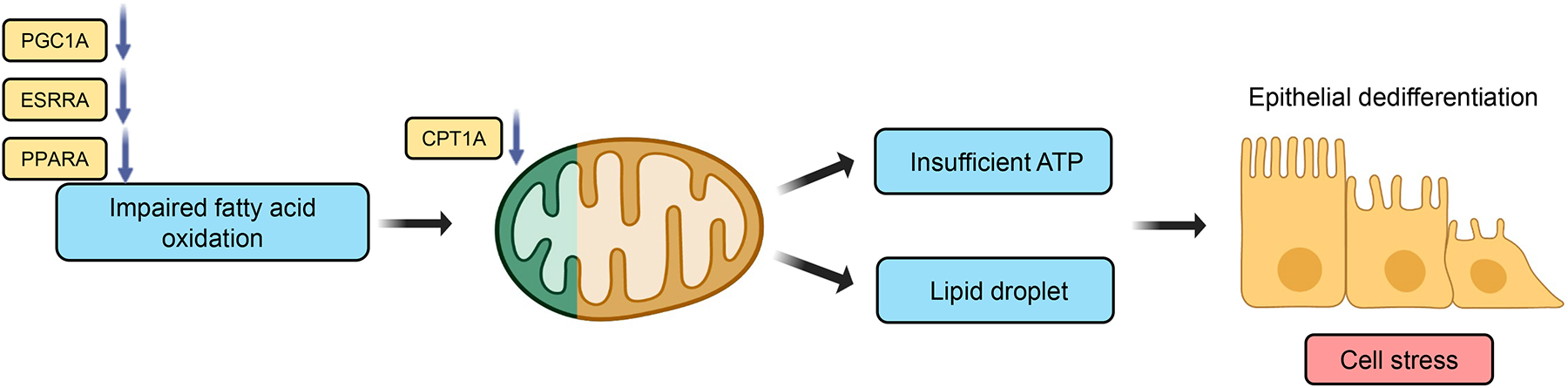
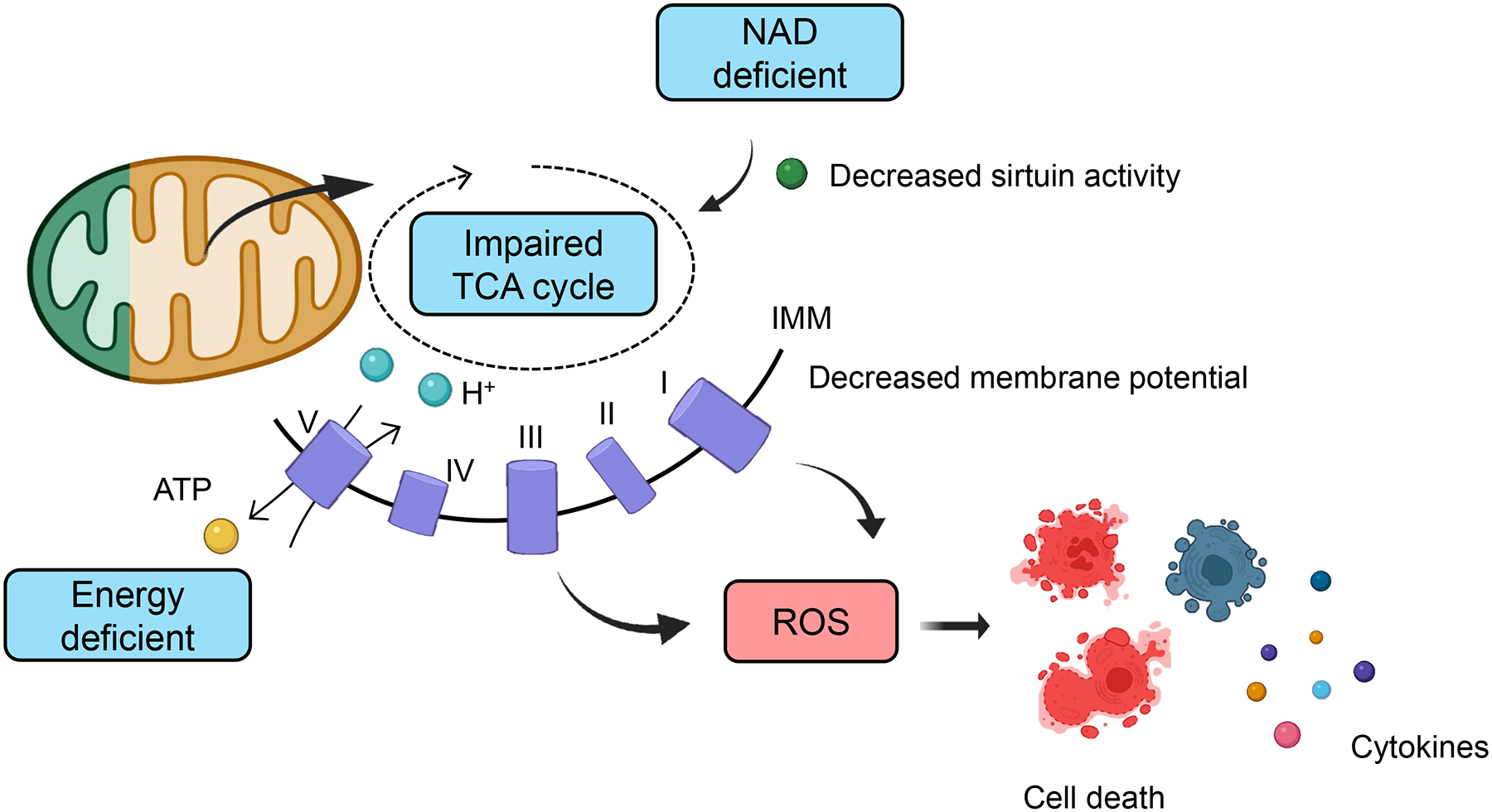
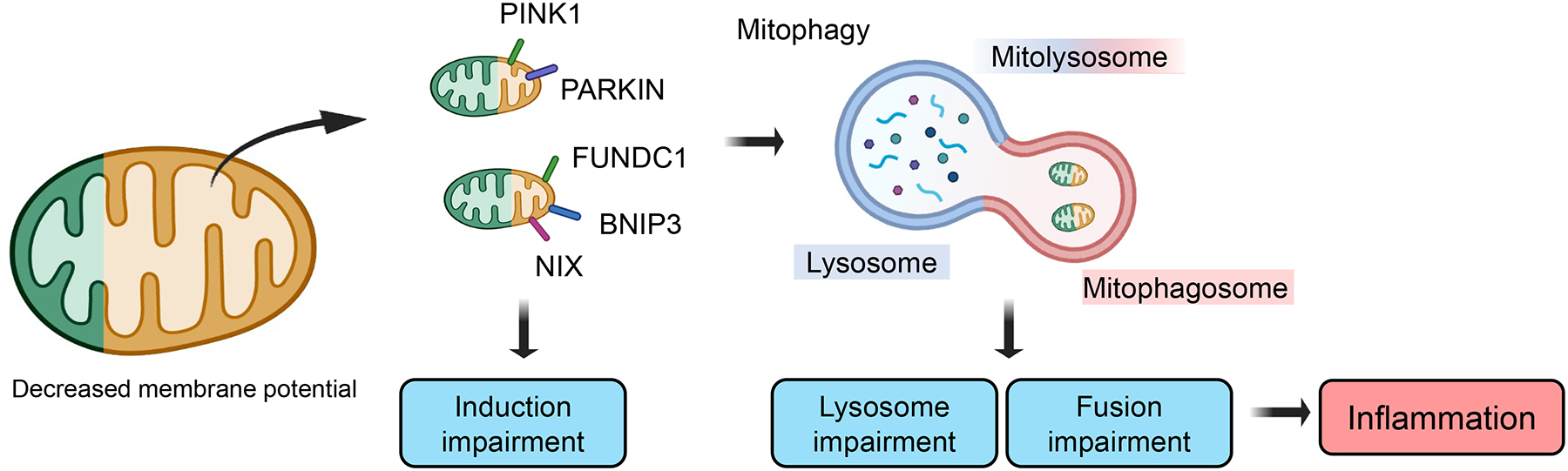
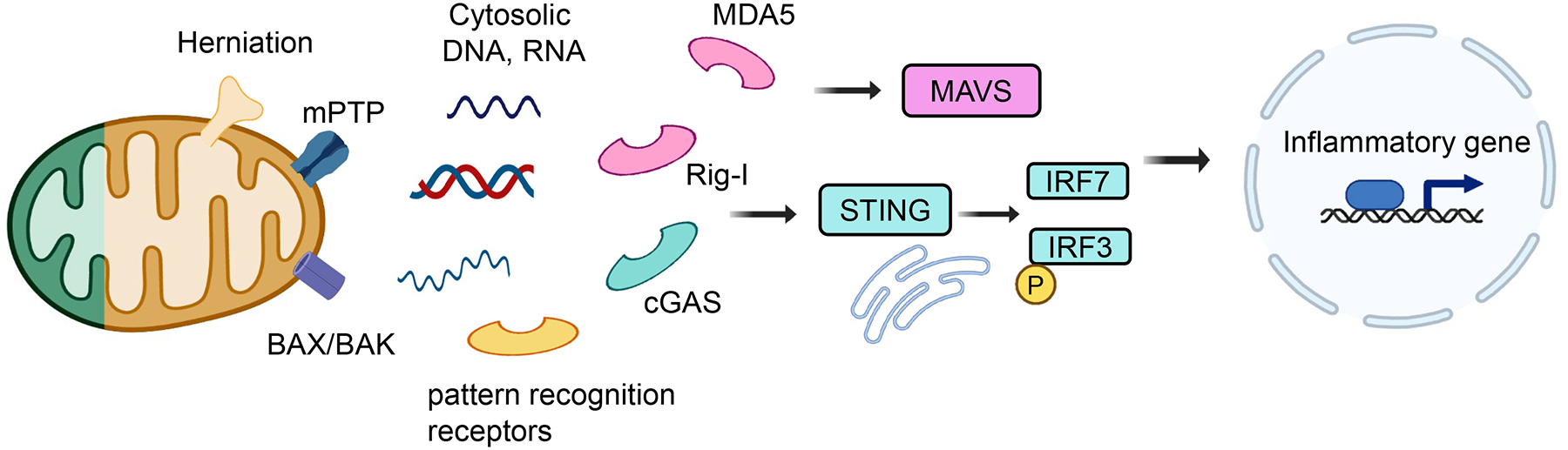
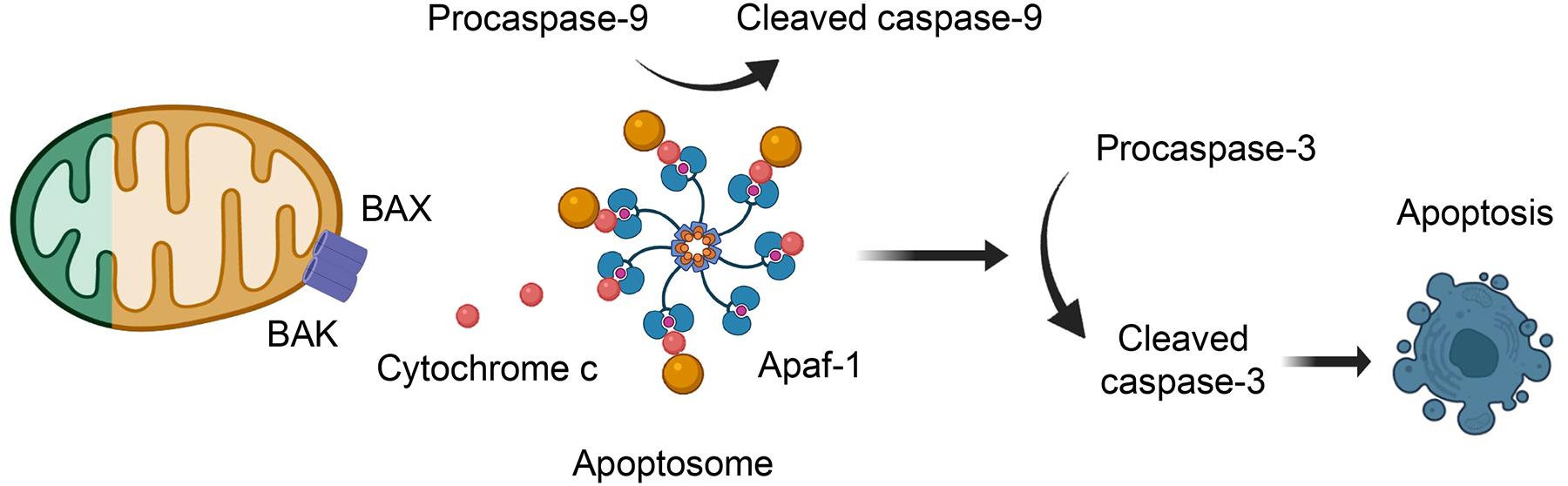
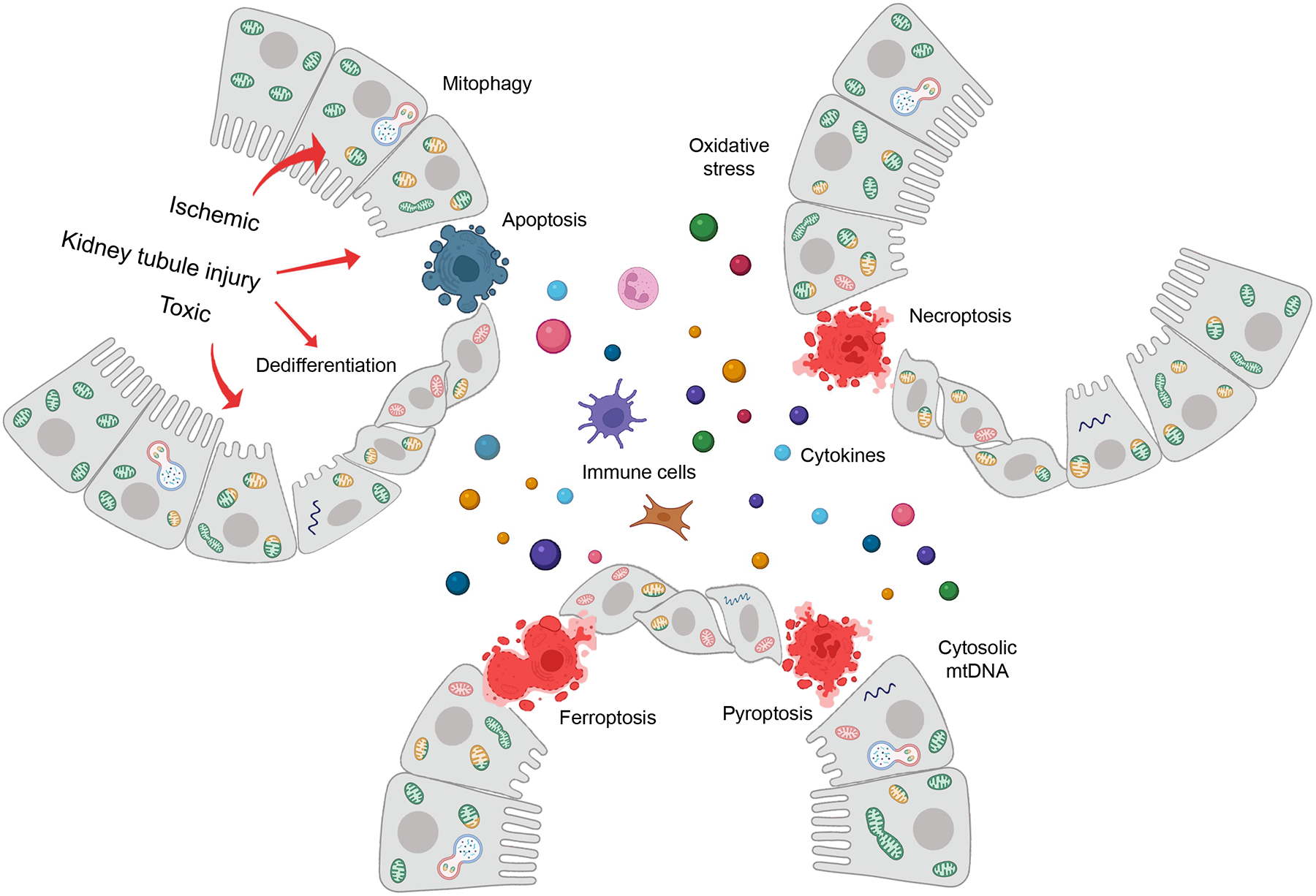
More than 800 million people suffer from kidney disease. Genetic studies and follow-up animal models and cell biological experiments indicate the key role of proximal tubule metabolism. Kidneys have one of the highest mitochondrial densities. Mitochondrial biogenesis, mitochondrial fusion and fission, and mitochondrial recycling, such as mitophagy are critical for proper mitochondrial function. Mitochondrial dysfunction can lead to an energetic crisis, orchestrate different types of cell death (apoptosis, necroptosis, pyroptosis, and ferroptosis), and influence cellular calcium levels and redox status. Collectively, mitochondrial defects in renal tubules contribute to epithelial atrophy, inflammation, or cell death, orchestrating kidney disease development.
Keywords: cell death; inflammation; kidney disease; mitochondria; mitophagy; renal tubule cell.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Similar articles
-
Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission.Redox Biol. 2020 Feb;30:101415. doi: 10.1016/j.redox.2019.101415. Epub 2019 Dec 28. Redox Biol. 2020. PMID: 31901590 Free PMC article.
-
A Molecular Approach to Mitophagy and Mitochondrial Dynamics.Mol Cells. 2018 Jan 31;41(1):18-26. doi: 10.14348/molcells.2018.2277. Epub 2018 Jan 23. Mol Cells. 2018. PMID: 29370689 Free PMC article. Review.
-
The Good and the Bad of Mitochondrial Breakups.Trends Cell Biol. 2019 Nov;29(11):888-900. doi: 10.1016/j.tcb.2019.08.003. Epub 2019 Sep 5. Trends Cell Biol. 2019. PMID: 31495461 Review.
-
UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury.J Pathol. 2019 Mar;247(3):392-405. doi: 10.1002/path.5198. Epub 2018 Dec 28. J Pathol. 2019. PMID: 30426490
-
Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury.Life Sci. 2019 Oct 15;235:116828. doi: 10.1016/j.lfs.2019.116828. Epub 2019 Aug 31. Life Sci. 2019. PMID: 31479679
Cited by
-
Drp1 Promotes Macrophage M1 Polarization and Inflammatory Response in Autoimmune Myocarditis by Driving Mitochondrial Fission.J Cardiovasc Transl Res. 2024 Oct 10. doi: 10.1007/s12265-024-10570-2. Online ahead of print. J Cardiovasc Transl Res. 2024. PMID: 39388091
-
Sacubitril/valsartan ameliorates tubulointerstitial fibrosis by restoring mitochondrial homeostasis in diabetic kidney disease.Diabetol Metab Syndr. 2024 Feb 10;16(1):40. doi: 10.1186/s13098-024-01284-1. Diabetol Metab Syndr. 2024. PMID: 38341600 Free PMC article.
-
Glutathione metabolism rewiring protects renal tubule cells against cisplatin-induced apoptosis and ferroptosis.Redox Rep. 2023 Dec;28(1):2152607. doi: 10.1080/13510002.2022.2152607. Redox Rep. 2023. PMID: 36692085 Free PMC article.
-
The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases.Int J Mol Sci. 2024 Jun 25;25(13):6936. doi: 10.3390/ijms25136936. Int J Mol Sci. 2024. PMID: 39000044 Free PMC article. Review.
-
Kidney Fibrosis and Oxidative Stress: From Molecular Pathways to New Pharmacological Opportunities.Biomolecules. 2024 Jan 22;14(1):137. doi: 10.3390/biom14010137. Biomolecules. 2024. PMID: 38275766 Free PMC article. Review.
References
-
- Zhong F et al. (2019) Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol 40 (12), 1120–1133. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
