

Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China
- PMID: 35467426
- PMCID: PMC9239062
- DOI: 10.1128/mbio.00463-22
Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China
Abstract
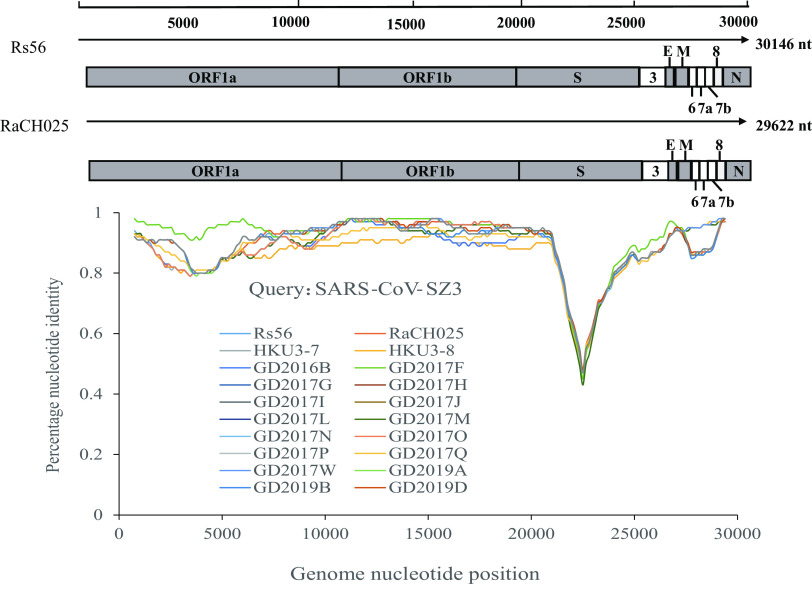
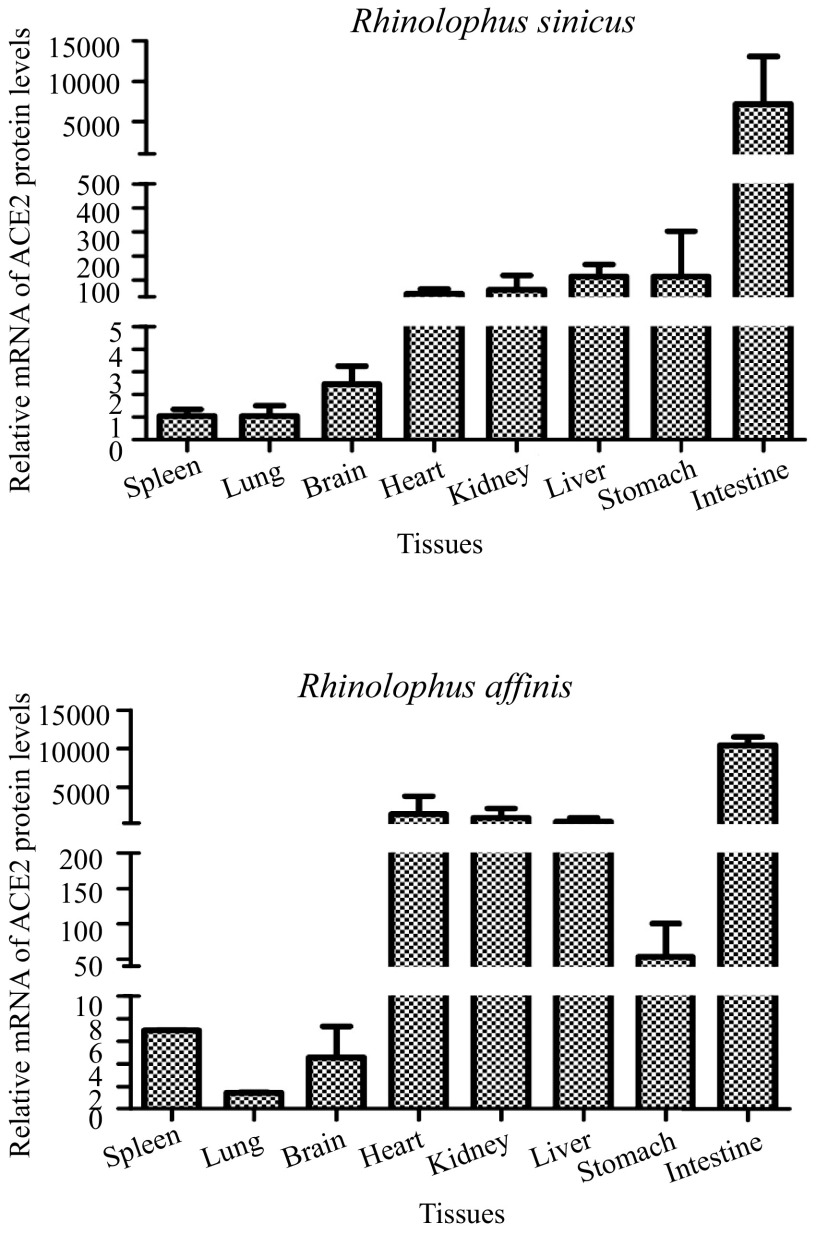
Severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) and SARS-CoV-2, the causative agents of SARS, which broke out in 2003, and coronavirus disease 2019 (COVID-2019), which broke out in 2019, probably originated in Rhinolophus sinicus and R. affinis, respectively. Rhinolophus bats are important hosts for coronaviruses. Many SARS-related coronaviruses (SARSr-CoVs) have been detected in bats from different areas of China; however, the diversity of bat SARSr-CoVs is increasing, and their transmission mechanisms have attracted much attention. Here, we report the findings of SARSr-CoVs in R. sinicus and R. affinis from South China from 2008 to 2021. The full-length genome sequences of the two novel SARSr-CoVs obtained from Guangdong shared 83 to 88% and 71 to 72% nucleotide identities with human SARS-CoV and SARS-CoV-2, respectively, while sharing high similarity with human SARS-CoV in hypervariable open reading frame 8 (ORF8). Significant recombination occurred between the two novel SARSr-CoVs. Phylogenetic analysis showed that the two novel bat SARSr-CoVs from Guangdong were more distant than the bat SARSr-CoVs from Yunnan to human SARS-CoV. We found that transmission in bats contributes more to virus diversity than time. Although our results of the sequence analysis of the receptor-binding motif (RBM) and the expression pattern of angiotensin-converting enzyme 2 (ACE2) inferred that these viruses could not directly infect humans, risks still exist after some unpredictable mutations. Thus, this study increased our understanding of the genetic diversity and transmission of SARSr-CoVs carried by bats in the field. IMPORTANCE Severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-CoV-2 probably originated from the SARS-related coronaviruses (SARSr-CoVs) carried by Rhinolophus bats from Yunnan, China. Systematic investigations of the reservoir hosts carrying SARSr-CoVs in Guangdong and the reservoir distribution and transmission are urgently needed to prevent future outbreaks. Here, we detected SARSr-CoV in Rhinolophus bat samples from Guangdong in 2009 and 2021 and found that the transmission of SARSr-CoV from different host populations contributes more to increased virus diversity than time. Bat SARSr-CoVs in Guangdong had genetic diversity, and Guangdong was also the hot spot for SARSr-CoVs. We once again prove that R. sinicus plays an important role in the maintenance of the SARS-CoVs. Besides, the SARSr-CoVs are mainly transmitted through the intestines in bats, and these SARSr-CoVs found in Guangdong could not use human ACE2 (hACE2), but whether they can pass through intermediate hosts or directly infect humans requires further research. Our findings demonstrate the ability of SARSr-CoVs to spread across species.
Keywords: SARS-related coronavirus; epidemiology; genetic diversity; horseshoe bats.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination.J Virol. 2015 Oct;89(20):10532-47. doi: 10.1128/JVI.01048-15. Epub 2015 Aug 12. J Virol. 2015. PMID: 26269185 Free PMC article.
-
Evolutionary Arms Race between Virus and Host Drives Genetic Diversity in Bat Severe Acute Respiratory Syndrome-Related Coronavirus Spike Genes.J Virol. 2020 Sep 29;94(20):e00902-20. doi: 10.1128/JVI.00902-20. Print 2020 Sep 29. J Virol. 2020. PMID: 32699095 Free PMC article.
-
Molecular epidemiology, evolution and phylogeny of SARS coronavirus.Infect Genet Evol. 2019 Jul;71:21-30. doi: 10.1016/j.meegid.2019.03.001. Epub 2019 Mar 4. Infect Genet Evol. 2019. PMID: 30844511 Free PMC article. Review.
-
Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events.J Virol. 2010 Mar;84(6):2808-19. doi: 10.1128/JVI.02219-09. Epub 2010 Jan 13. J Virol. 2010. PMID: 20071579 Free PMC article.
-
Geographical structure of bat SARS-related coronaviruses.Infect Genet Evol. 2019 Apr;69:224-229. doi: 10.1016/j.meegid.2019.02.001. Epub 2019 Feb 6. Infect Genet Evol. 2019. PMID: 30735813 Free PMC article. Review.
Cited by
-
Identification of a novel adenovirus in liver tissue sample of the Great Himalayan leaf-nosed bat (Hipposideros armiger).Braz J Microbiol. 2024 Mar;55(1):117-123. doi: 10.1007/s42770-024-01258-5. Epub 2024 Jan 23. Braz J Microbiol. 2024. PMID: 38261263
-
A comprehensive dataset of animal-associated sarbecoviruses.Sci Data. 2023 Oct 7;10(1):681. doi: 10.1038/s41597-023-02558-5. Sci Data. 2023. PMID: 37805633 Free PMC article.
-
Genotype and Phenotype Characterization of Rhinolophus sp. Sarbecoviruses from Vietnam: Implications for Coronavirus Emergence.Viruses. 2023 Sep 8;15(9):1897. doi: 10.3390/v15091897. Viruses. 2023. PMID: 37766303 Free PMC article.
-
Rhinolophus sinicus virome revealed multiple novel mosquito-borne zoonotic viruses.Front Cell Infect Microbiol. 2022 Oct 11;12:960507. doi: 10.3389/fcimb.2022.960507. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 36304937 Free PMC article.
References
-
- Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JSM, Poon LLM. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278. doi:10.1126/science.1087139. - DOI - PubMed
-
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi:10.1038/s41586-020-2012-7. - DOI - PMC - PubMed
-
- Zhu N, Zhang DY, Wang WL, Li XW, Yang B, Song JD, Zhao X, Huang BY, Shi WF, Lu RJ, Niu PH, Zhan FX, Ma XJ, Wang DY, Xu WB, Wu GZ, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team . 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. doi:10.1056/NEJMoa2001017. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
