

Impact of DNA damage response defects in cancer cells on response to immunotherapy and radiotherapy
- PMID: 35460184
- PMCID: PMC9321602
- DOI: 10.1111/1754-9485.13413
Impact of DNA damage response defects in cancer cells on response to immunotherapy and radiotherapy
Abstract
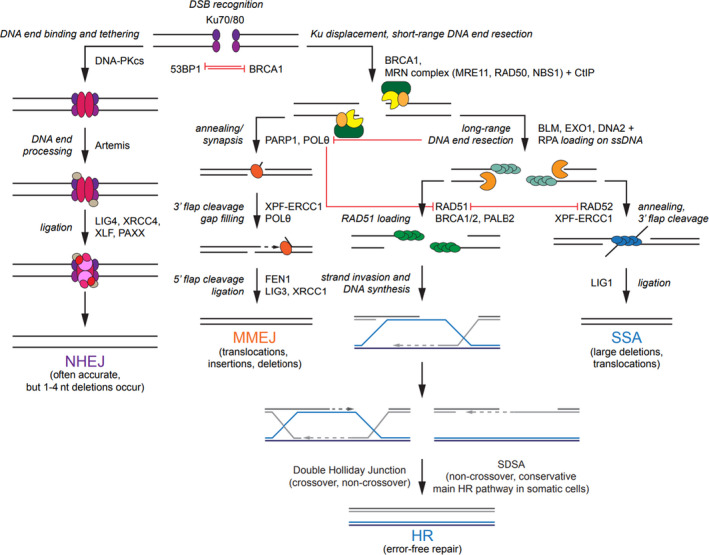
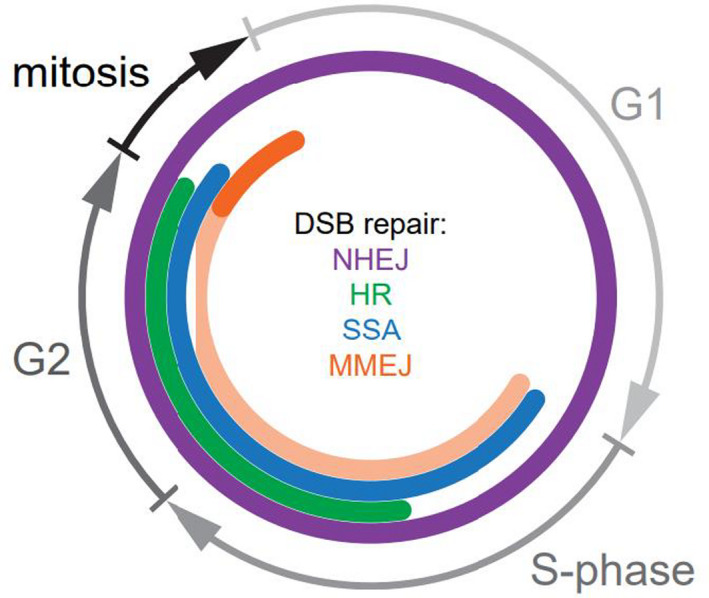
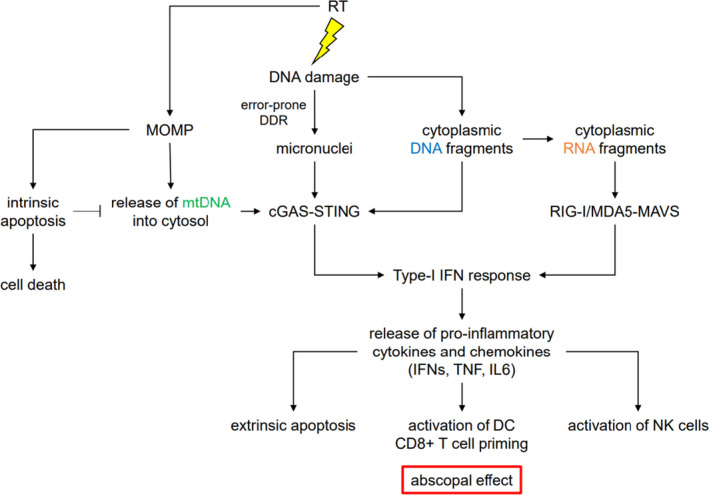
The DNA damage response (DDR) is a complex set of downstream pathways triggered in response to DNA damage to maintain genomic stability. Many tumours exhibit mutations which inactivate components of the DDR, making them prone to the accumulation of DNA defects. These can both facilitate the development of tumours and provide potential targets for novel therapeutic interventions. The inhibition of the DDR has been shown to induce radiosensitivity in certain cancers, rendering them susceptible to treatment with radiotherapy and improving the therapeutic window. Moreover, DDR defects are a strong predictor of patient response to immune checkpoint inhibition (ICI). The ability to target the DDR selectively has the potential to expand the tumour neoantigen repertoire, thus increasing tumour immunogenicity and facilitating a CD8+ T and NK cell response against cancer cells. Combinatorial approaches, which seek to integrate DDR inhibition with radiotherapy and immunotherapy, have shown promise in early trials. Further studies are necessary to understand these synergies and establish reliable biomarkers.
Keywords: DNA damage response; cancer; double-strand breaks; immunotherapy; radiotherapy.
© 2022 The Authors. Journal of Medical Imaging and Radiation Oncology published by John Wiley & Sons Australia, Ltd on behalf of Royal Australian and New Zealand College of Radiologists.
Figures
Similar articles
-
The complementarity of DDR, nucleic acids and anti-tumour immunity.Nature. 2023 Jul;619(7970):475-486. doi: 10.1038/s41586-023-06069-6. Epub 2023 Jul 19. Nature. 2023. PMID: 37468584 Review.
-
Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy.Front Immunol. 2021 Dec 14;12:797880. doi: 10.3389/fimmu.2021.797880. eCollection 2021. Front Immunol. 2021. PMID: 34970273 Free PMC article. Review.
-
Diverse immune response of DNA damage repair-deficient tumors.Cell Rep Med. 2021 May 18;2(5):100276. doi: 10.1016/j.xcrm.2021.100276. eCollection 2021 May 18. Cell Rep Med. 2021. PMID: 34095878 Free PMC article.
-
Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy.Int J Mol Sci. 2022 Mar 17;23(6):3238. doi: 10.3390/ijms23063238. Int J Mol Sci. 2022. PMID: 35328658 Free PMC article. Review.
-
Analysis of DNA Damage Response Gene Alterations and Tumor Mutational Burden Across 17,486 Tubular Gastrointestinal Carcinomas: Implications for Therapy.Oncologist. 2019 Oct;24(10):1340-1347. doi: 10.1634/theoncologist.2019-0034. Epub 2019 Apr 30. Oncologist. 2019. PMID: 31040255 Free PMC article.
Cited by
-
Epigenetic modification in radiotherapy and immunotherapy for cancers.Tzu Chi Med J. 2024 Sep 5;36(4):396-406. doi: 10.4103/tcmj.tcmj_3_24. eCollection 2024 Oct-Dec. Tzu Chi Med J. 2024. PMID: 39421493 Free PMC article. Review.
-
Analysis of Tumor Mutational Burden, Progression-Free Survival, and Local-Regional Control in Patents with Locally Advanced Non-Small Cell Lung Cancer Treated With Chemoradiation and Durvalumab.JAMA Netw Open. 2023 Jan 3;6(1):e2249591. doi: 10.1001/jamanetworkopen.2022.49591. JAMA Netw Open. 2023. PMID: 36602799 Free PMC article.
-
Metal nanoparticles for cancer therapy: Precision targeting of DNA damage.Acta Pharm Sin B. 2024 Mar;14(3):1132-1149. doi: 10.1016/j.apsb.2023.08.031. Epub 2023 Sep 3. Acta Pharm Sin B. 2024. PMID: 38486992 Free PMC article. Review.
-
Nanoparticle-Mediated Radiotherapy: Unraveling Dose Enhancement and Apoptotic Responses in Cancer and Normal Cell Lines.Biomolecules. 2023 Nov 29;13(12):1720. doi: 10.3390/biom13121720. Biomolecules. 2023. PMID: 38136591 Free PMC article.
-
DNA Damage and Its Role in Cancer Therapeutics.Int J Mol Sci. 2023 Mar 1;24(5):4741. doi: 10.3390/ijms24054741. Int J Mol Sci. 2023. PMID: 36902170 Free PMC article. Review.
References
-
- Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, Postel‐Vinay S. Targeting the DNA damage response in immuno‐oncology: developments and opportunities. Nat Rev Cancer 2021; 21: 701–17. - PubMed
-
- Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 2016; 16: 35–42. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
