

Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives
- PMID: 35454131
- PMCID: PMC9030615
- DOI: 10.3390/biom12040542
Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives
Abstract
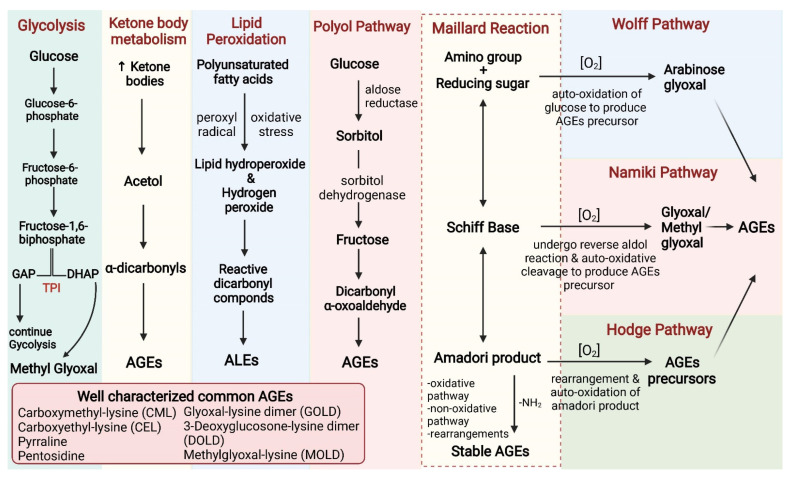
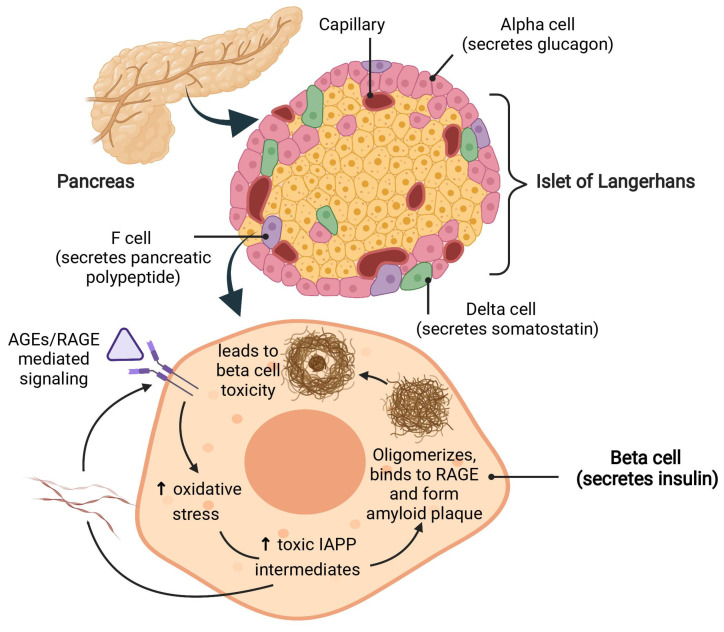
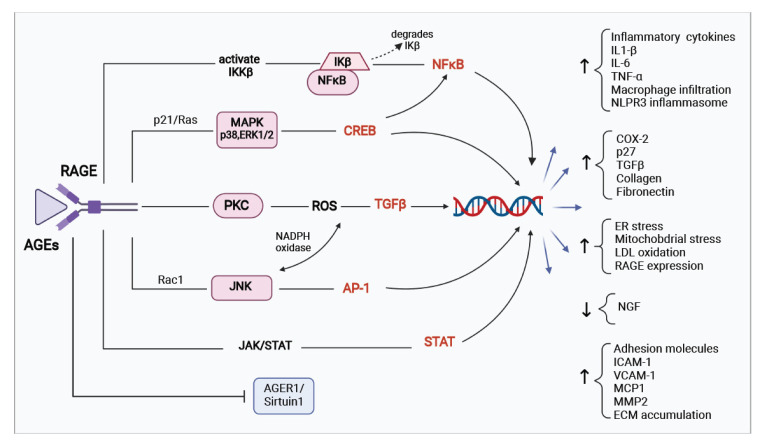
Persistent hyperglycemic state in type 2 diabetes mellitus leads to the initiation and progression of non-enzymatic glycation reaction with proteins and lipids and nucleic acids. Glycation reaction leads to the generation of a heterogeneous group of chemical moieties known as advanced glycated end products (AGEs), which play a central role in the pathophysiology of diabetic complications. The engagement of AGEs with its chief cellular receptor, RAGE, activates a myriad of signaling pathways such as MAPK/ERK, TGF-β, JNK, and NF-κB, leading to enhanced oxidative stress and inflammation. The downstream consequences of the AGEs/RAGE axis involve compromised insulin signaling, perturbation of metabolic homeostasis, RAGE-induced pancreatic beta cell toxicity, and epigenetic modifications. The AGEs/RAGE signaling instigated modulation of gene transcription is profoundly associated with the progression of type 2 diabetes mellitus and pathogenesis of diabetic complications. In this review, we will summarize the exogenous and endogenous sources of AGEs, their role in metabolic dysfunction, and current understandings of AGEs/RAGE signaling cascade. The focus of this review is to recapitulate the role of the AGEs/RAGE axis in the pathogenesis of type 2 diabetes mellitus and its associated complications. Furthermore, we present an overview of future perspectives to offer new therapeutic interventions to intervene with the AGEs/RAGE signaling pathway and to slow down the progression of diabetes-related complications.
Keywords: advanced glycation end products (AGEs); diabetic complications; hyperglycemia; pancreatic beta cells; receptor for advanced glycation end products (RAGE); type 2 diabetes mellitus.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures
Similar articles
-
Mechanistic role of Syzygium cumini (L.) Skeels in glycation induced diabetic nephropathy via RAGE-NF-κB pathway and extracellular proteins modifications: A molecular approach.J Ethnopharmacol. 2024 Mar 25;322:117573. doi: 10.1016/j.jep.2023.117573. Epub 2023 Dec 16. J Ethnopharmacol. 2024. PMID: 38110133
-
SiJunZi decoction ameliorates bone quality and redox homeostasis and regulates advanced glycation end products/receptor for advanced glycation end products and WNT/β-catenin signaling pathways in diabetic mice.J Ethnopharmacol. 2024 Jan 30;319(Pt 2):117167. doi: 10.1016/j.jep.2023.117167. Epub 2023 Sep 14. J Ethnopharmacol. 2024. PMID: 37716489
-
Action of metformin therapy against advanced glycation, oxidative stress and inflammation in type 2 diabetes patients: 3 months follow-up study.Diabetes Metab Syndr. 2020 Sep-Oct;14(5):1449-1458. doi: 10.1016/j.dsx.2020.07.036. Epub 2020 Jul 28. Diabetes Metab Syndr. 2020. PMID: 32769032
-
The dynamic roles of advanced glycation end products.Vitam Horm. 2024;125:1-29. doi: 10.1016/bs.vh.2024.02.008. Epub 2024 May 28. Vitam Horm. 2024. PMID: 38997161 Review.
-
AGE-RAGE axis blockade in diabetic nephropathy: Current status and future directions.Eur J Pharmacol. 2018 Aug 15;833:158-164. doi: 10.1016/j.ejphar.2018.06.001. Epub 2018 Jun 5. Eur J Pharmacol. 2018. PMID: 29883668 Review.
Cited by
-
Polysaccharides from Seedless Chestnut Rose (Rosa sterilis) Fruits: Insights into Innovative Drying Technologies and Their Structural Characteristics, Antioxidant, Antiglycation, and α-Glucosidase Inhibitory Activities.Foods. 2024 Aug 7;13(16):2483. doi: 10.3390/foods13162483. Foods. 2024. PMID: 39200410 Free PMC article.
-
Serum Pentosidine is Associated with Cardiac Dysfunction and Atherosclerosis in T2DM.Diabetes Metab Syndr Obes. 2023 Jan 26;16:237-244. doi: 10.2147/DMSO.S398119. eCollection 2023. Diabetes Metab Syndr Obes. 2023. PMID: 36760597 Free PMC article.
-
Multi-omics analysis reveals attenuation of cellular stress by empagliflozin in high glucose-treated human cardiomyocytes.J Transl Med. 2023 Sep 23;21(1):662. doi: 10.1186/s12967-023-04537-1. J Transl Med. 2023. PMID: 37742032 Free PMC article.
-
A Narrative Review of the Advances in Screening Methods for Diabetic Retinopathy: Enhancing Early Detection and Vision Preservation.Cureus. 2024 Feb 4;16(2):e53586. doi: 10.7759/cureus.53586. eCollection 2024 Feb. Cureus. 2024. PMID: 38455792 Free PMC article. Review.
-
Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease.Antioxidants (Basel). 2024 Apr 12;13(4):455. doi: 10.3390/antiox13040455. Antioxidants (Basel). 2024. PMID: 38671903 Free PMC article. Review.
References
-
- Mohammedi K., Woodward M., Marre M., Colagiuri S., Cooper M., Harrap S., Mancia G., Poulter N., Williams B., Zoungas S. Comparative Effects of Microvascular and Macrovascular Disease on the Risk of Major Outcomes in Patients with Type 2 Diabetes. Cardiovasc. Diabetol. 2017;16:95. doi: 10.1186/s12933-017-0574-y. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
