

Structural basis of protein substrate processing by human mitochondrial high-temperature requirement A2 protease
- PMID: 35452308
- PMCID: PMC9170070
- DOI: 10.1073/pnas.2203172119
Structural basis of protein substrate processing by human mitochondrial high-temperature requirement A2 protease
Abstract
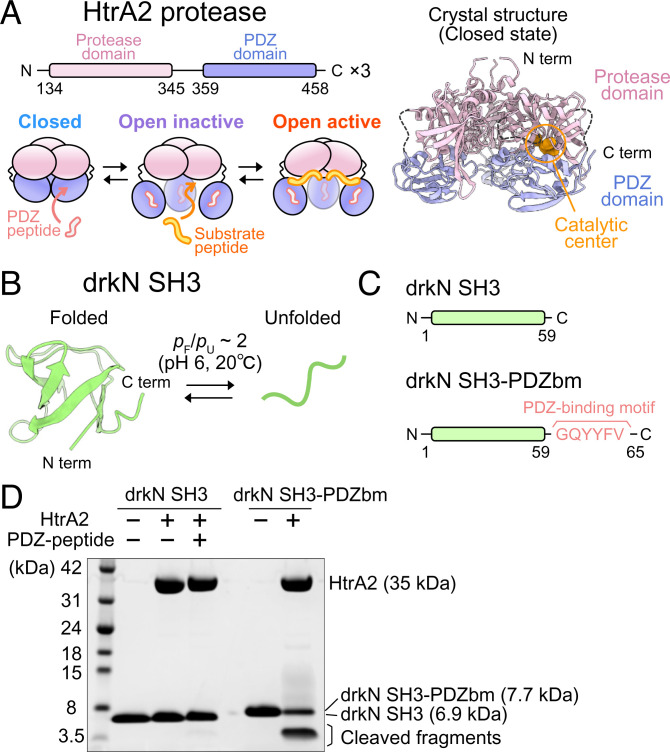
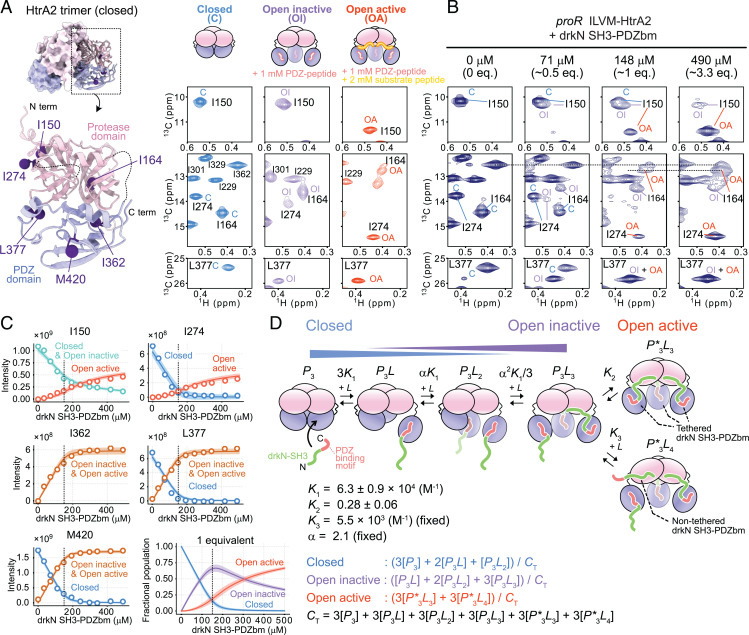
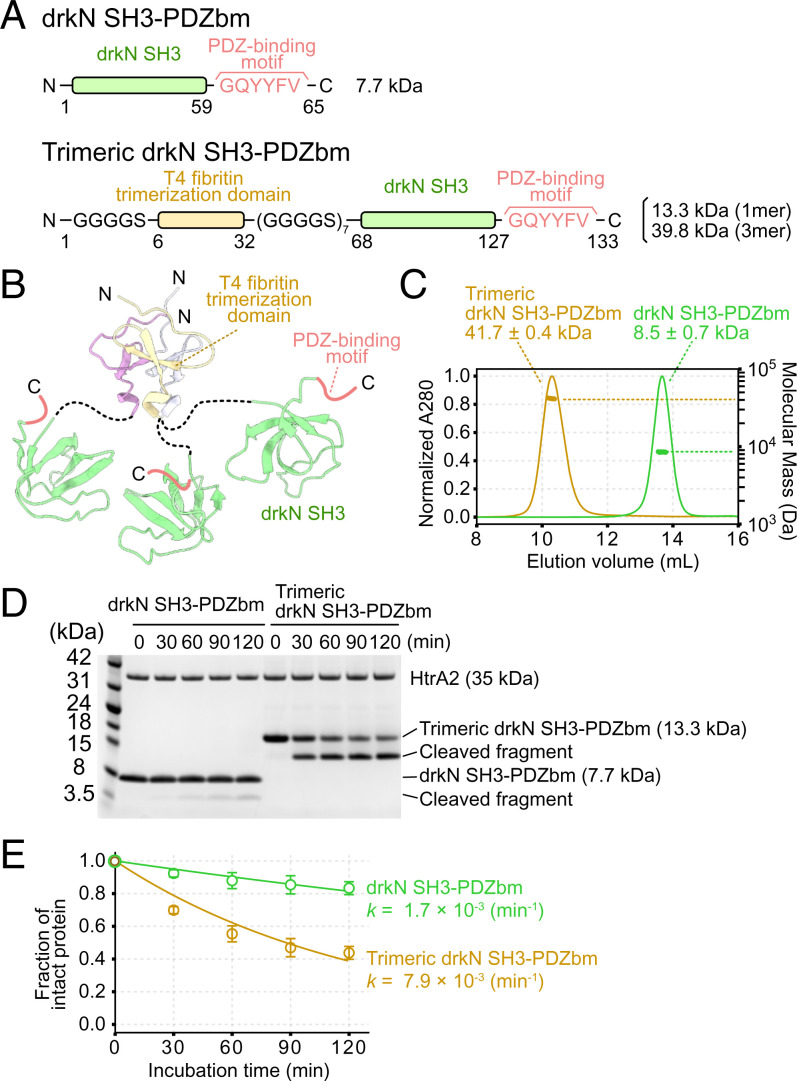
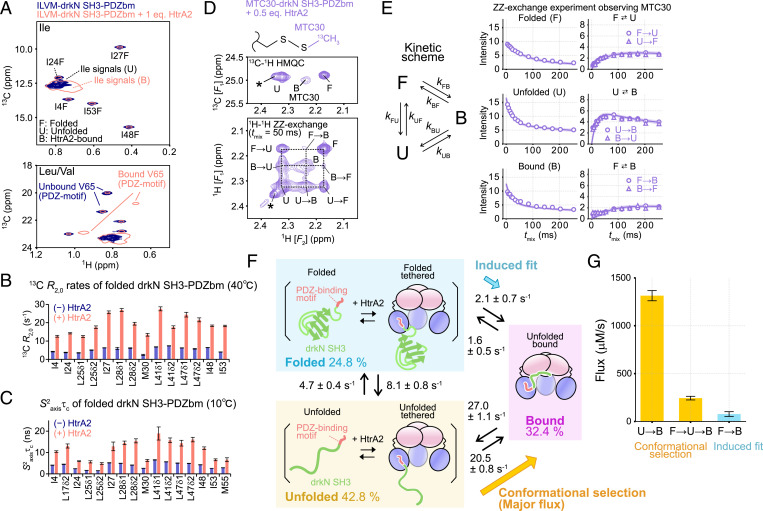
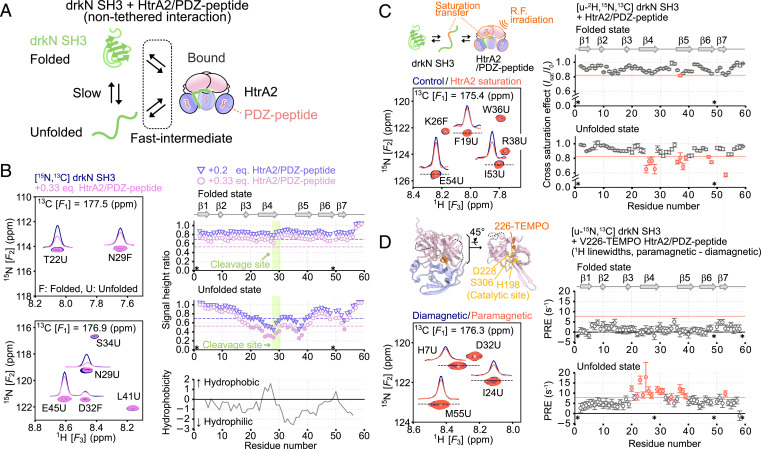
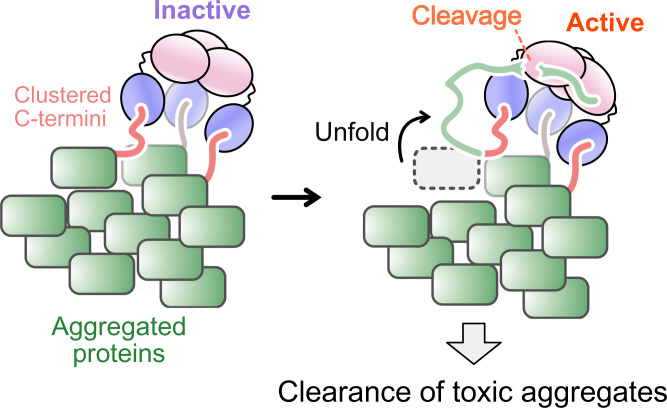
The human high-temperature requirement A2 (HtrA2) protein is a trimeric protease that cleaves misfolded proteins to protect cells from stresses caused by toxic, proteinaceous aggregates, and the aberrant function of HtrA2 is closely related to the onset of neurodegenerative disorders. Our methyl-transverse relaxation optimized spectroscopy (TROSY)–based NMR studies using small-peptide ligands have previously revealed a stepwise activation mechanism involving multiple distinct conformational states. However, very little is known about how HtrA2 binds to protein substrates and if the distinct conformational states observed in previous peptide studies might be involved in the processing of protein clients. Herein, we use solution-based NMR spectroscopy to investigate the interaction between the N-terminal Src homology 3 domain from downstream of receptor kinase (drk) with an added C-terminal HtrA2-binding motif (drkN SH3-PDZbm) that exhibits marginal folding stability and serves as a mimic of a physiological protein substrate. We show that drkN SH3-PDZbm binds to HtrA2 via a two-pronged interaction, involving both its C-terminal PDZ-domain binding motif and a central hydrophobic region, with binding occurring preferentially via an unfolded ensemble of substrate molecules. Multivalent interactions between several clients and a single HtrA2 trimer significantly stimulate the catalytic activity of HtrA2, suggesting that binding avidity plays an important role in regulating substrate processing. Our results provide a thermodynamic, kinetic, and structural description of the interaction of HtrA2 with protein substrates and highlight the importance of a trimeric architecture for function as a stress-protective protease that mitigates aggregation.
Keywords: conformational selection; ligand-binding thermodynamics; methyl transverse relaxation optimized NMR spectroscopy; mitochondrial proteostasis; protein–protein interaction.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
Oligomeric assembly regulating mitochondrial HtrA2 function as examined by methyl-TROSY NMR.Proc Natl Acad Sci U S A. 2021 Mar 16;118(11):e2025022118. doi: 10.1073/pnas.2025022118. Proc Natl Acad Sci U S A. 2021. PMID: 33692127 Free PMC article.
-
Dissecting the role of interprotomer cooperativity in the activation of oligomeric high-temperature requirement A2 protein.Proc Natl Acad Sci U S A. 2021 Aug 31;118(35):e2111257118. doi: 10.1073/pnas.2111257118. Proc Natl Acad Sci U S A. 2021. PMID: 34446566 Free PMC article.
-
Temperature-induced changes of HtrA2(Omi) protease activity and structure.Cell Stress Chaperones. 2013 Jan;18(1):35-51. doi: 10.1007/s12192-012-0355-1. Epub 2012 Aug 1. Cell Stress Chaperones. 2013. PMID: 22851136 Free PMC article.
-
Unraveling the Dichotomy of Enigmatic Serine Protease HtrA2.Front Mol Biosci. 2022 Feb 3;9:824846. doi: 10.3389/fmolb.2022.824846. eCollection 2022. Front Mol Biosci. 2022. PMID: 35187085 Free PMC article. Review.
-
Pathogenic Role of Serine Protease HtrA2/Omi in Neurodegenerative Diseases.Curr Protein Pept Sci. 2017;18(7):746-757. doi: 10.2174/1389203717666160311115750. Curr Protein Pept Sci. 2017. PMID: 26965693 Review.
Cited by
-
Synergism between x-ray crystallography and NMR residual dipolar couplings in characterizing protein dynamics.Struct Dyn. 2023 Jul 11;10(4):040901. doi: 10.1063/4.0000192. eCollection 2023 Jul. Struct Dyn. 2023. PMID: 37448874 Free PMC article.
-
α-Synuclein and Mitochondria: Probing the Dynamics of Disordered Membrane-protein Regions Using Solid-State Nuclear Magnetic Resonance.JACS Au. 2024 May 28;4(6):2372-2380. doi: 10.1021/jacsau.4c00323. eCollection 2024 Jun 24. JACS Au. 2024. PMID: 38938811 Free PMC article.
-
HTRA2/OMI-Mediated Mitochondrial Quality Control Alters Macrophage Polarization Affecting Systemic Chronic Inflammation.Int J Mol Sci. 2024 Jan 27;25(3):1577. doi: 10.3390/ijms25031577. Int J Mol Sci. 2024. PMID: 38338855 Free PMC article. Review.
-
Genetic variations as predictors of dispositional and dyadic empathy-a couple study.Sci Rep. 2024 Nov 9;14(1):27411. doi: 10.1038/s41598-024-78857-7. Sci Rep. 2024. PMID: 39521899 Free PMC article.
-
The Repeating, Modular Architecture of the HtrA Proteases.Biomolecules. 2022 Jun 7;12(6):793. doi: 10.3390/biom12060793. Biomolecules. 2022. PMID: 35740918 Free PMC article.
References
-
- Dobson C. M., Getting out of shape. Nature 418, 729–730 (2002). - PubMed
-
- Gregersen N., Bross P., Vang S., Christensen J. H., Protein misfolding and human disease. Annu. Rev. Genomics Hum. Genet. 7, 103–124 (2006). - PubMed
-
- Chiti F., Dobson C. M., Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006). - PubMed
-
- Clausen T., Kaiser M., Huber R., Ehrmann M., HTRA proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 12, 152–162 (2011). - PubMed
-
- Skorko-Glonek J., et al. , HtrA protease family as therapeutic targets. Curr. Pharm. Des. 19, 977–1009 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
