

NMR Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins
- PMID: 35446534
- PMCID: PMC9136928
- DOI: 10.1021/acs.chemrev.1c01023
NMR Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins
Abstract
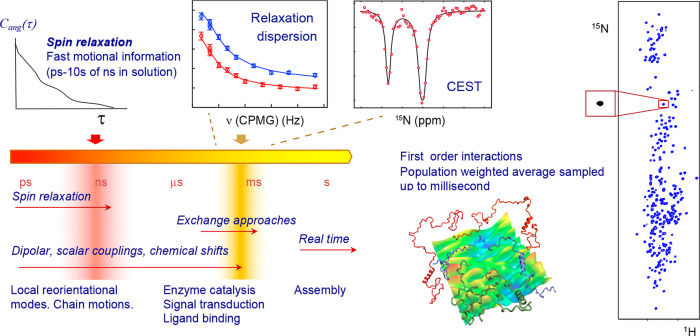
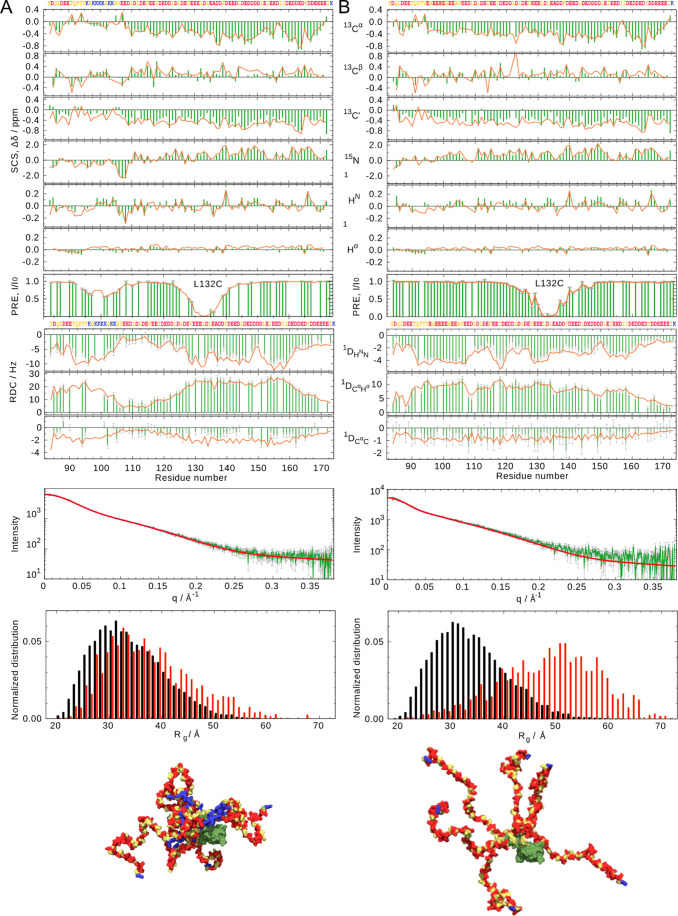
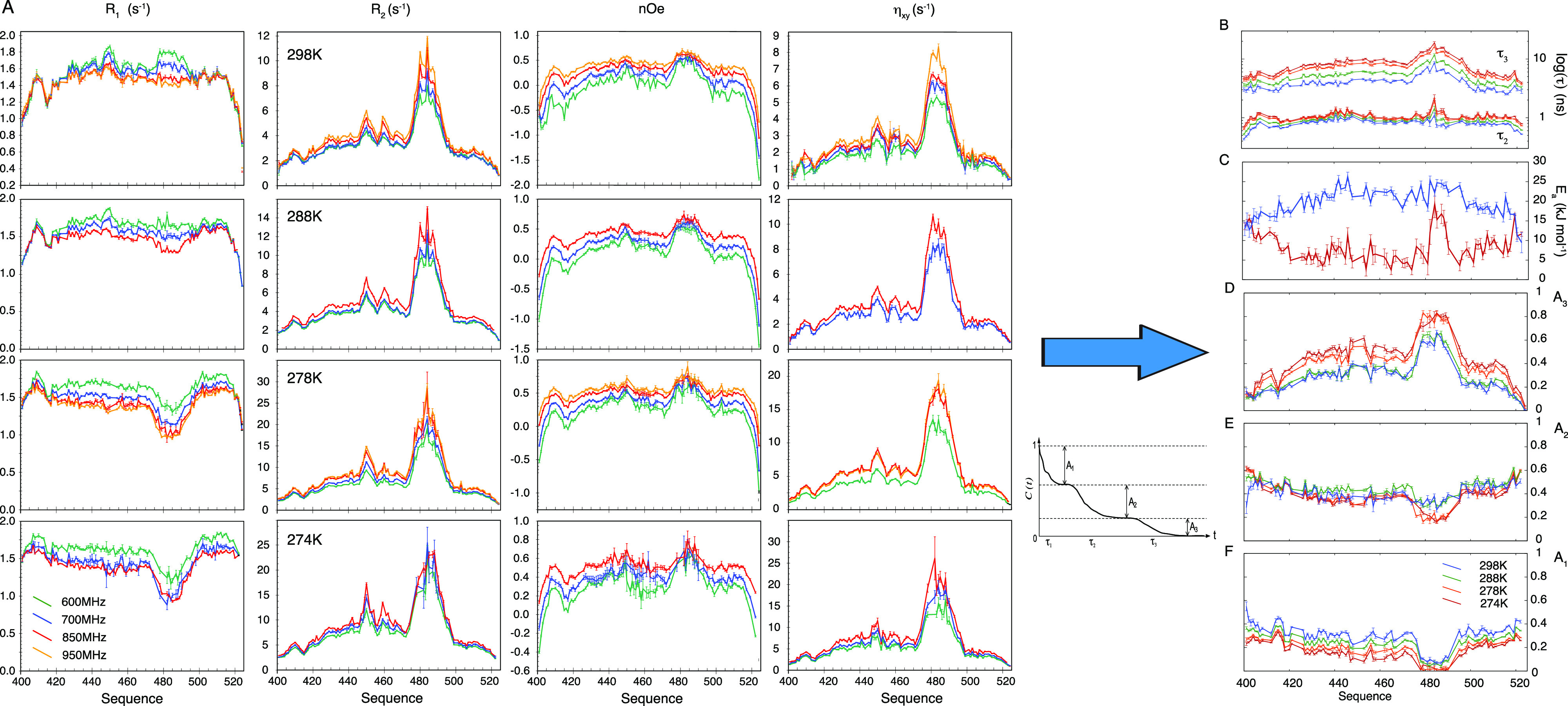
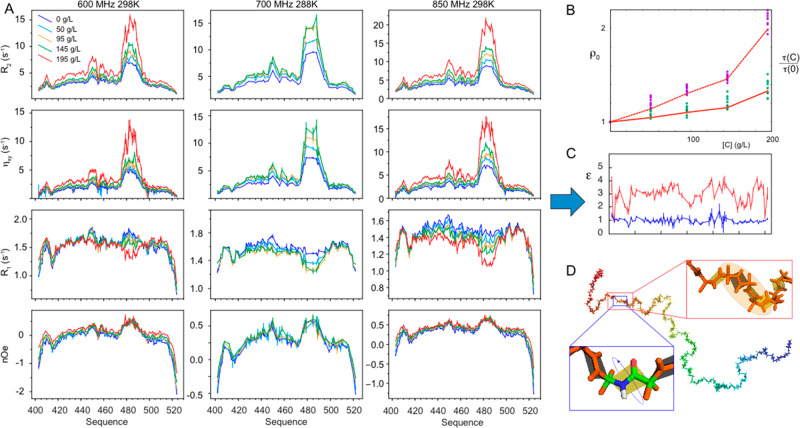
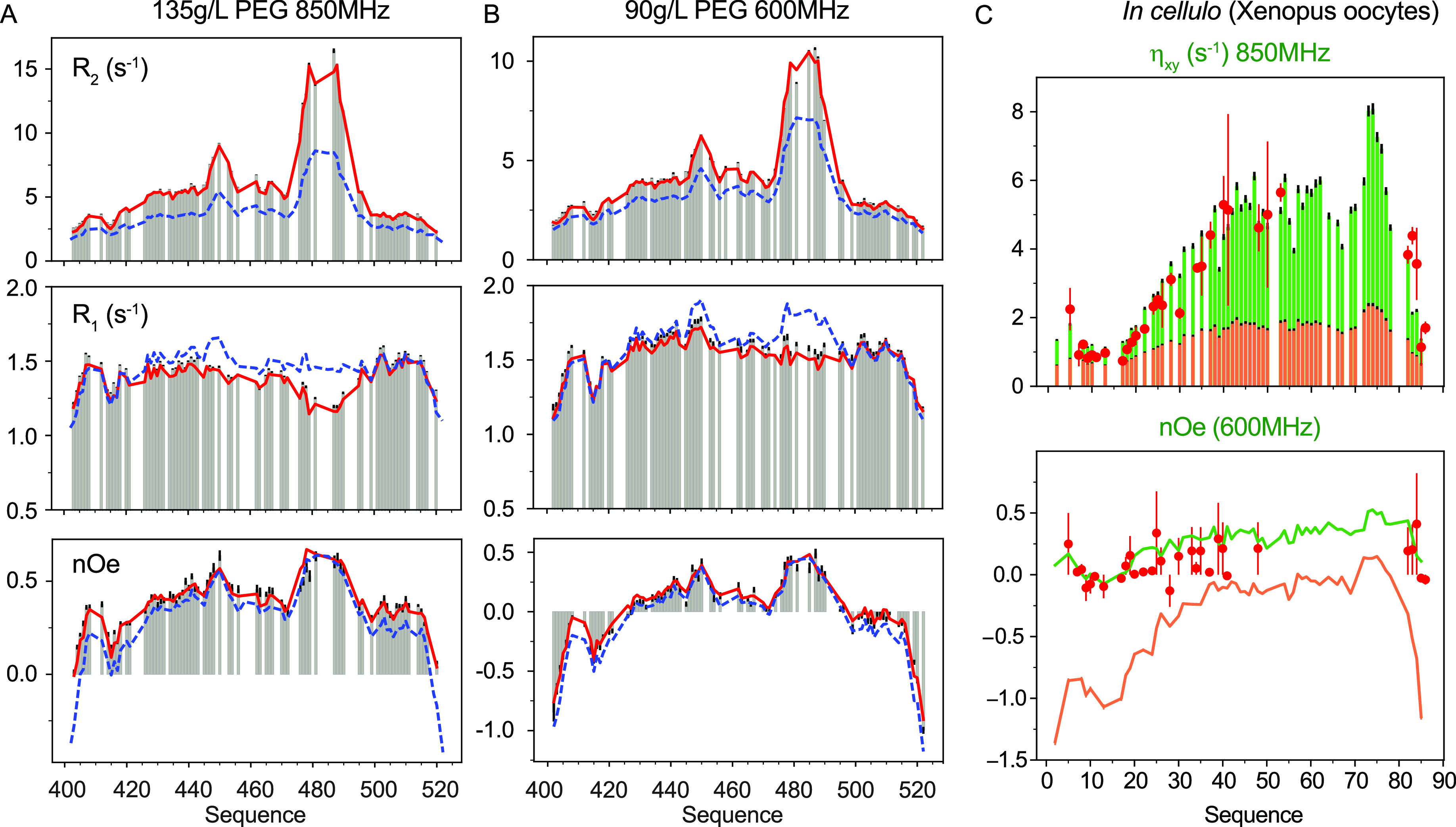
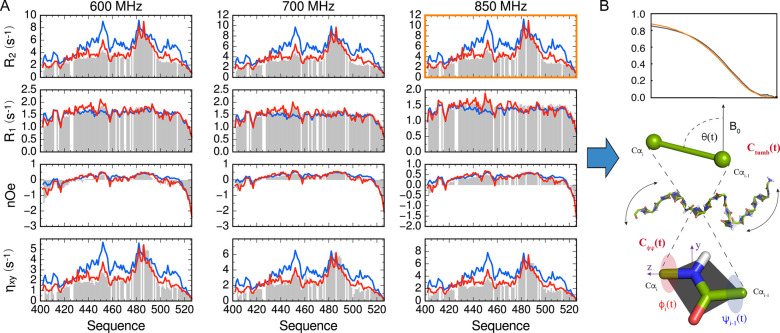
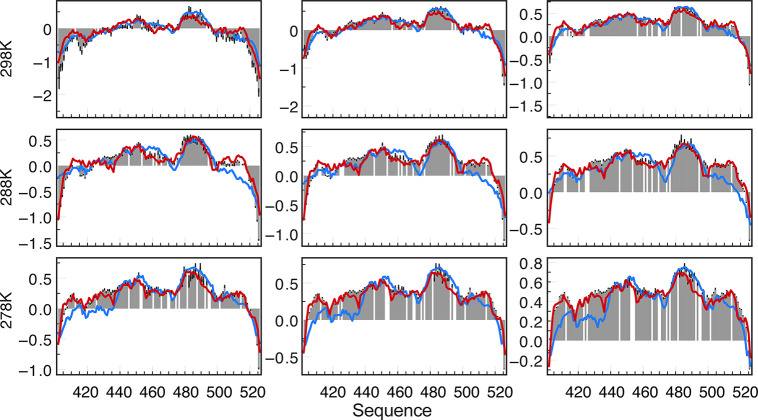
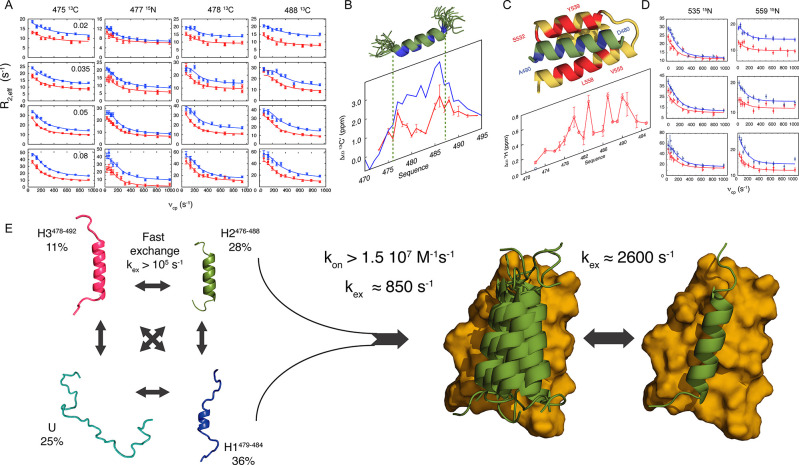
Intrinsically disordered proteins are ubiquitous throughout all known proteomes, playing essential roles in all aspects of cellular and extracellular biochemistry. To understand their function, it is necessary to determine their structural and dynamic behavior and to describe the physical chemistry of their interaction trajectories. Nuclear magnetic resonance is perfectly adapted to this task, providing ensemble averaged structural and dynamic parameters that report on each assigned resonance in the molecule, unveiling otherwise inaccessible insight into the reaction kinetics and thermodynamics that are essential for function. In this review, we describe recent applications of NMR-based approaches to understanding the conformational energy landscape, the nature and time scales of local and long-range dynamics and how they depend on the environment, even in the cell. Finally, we illustrate the ability of NMR to uncover the mechanistic basis of functional disordered molecular assemblies that are important for human health.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
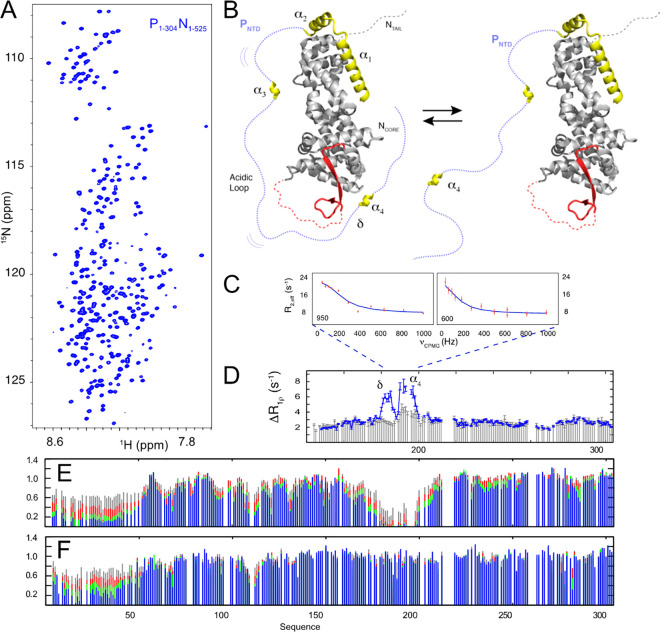
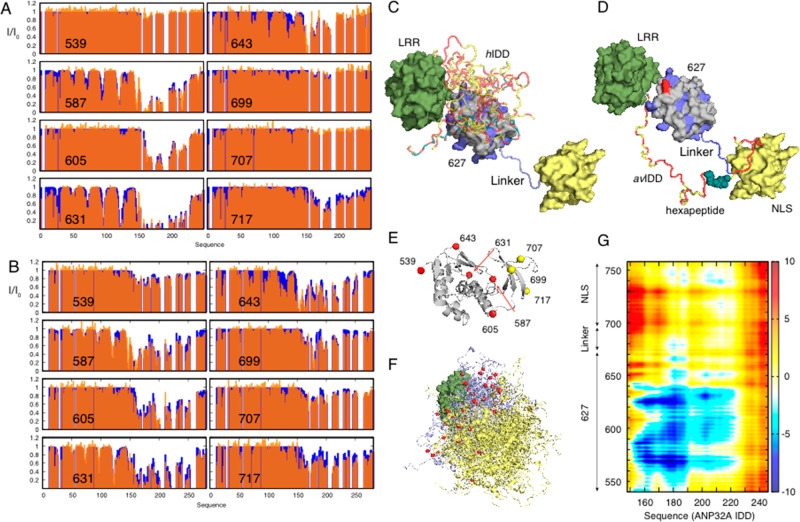
Similar articles
-
Atomic resolution conformational dynamics of intrinsically disordered proteins from NMR spin relaxation.Prog Nucl Magn Reson Spectrosc. 2017 Nov;102-103:43-60. doi: 10.1016/j.pnmrs.2017.06.001. Epub 2017 Jul 10. Prog Nucl Magn Reson Spectrosc. 2017. PMID: 29157493 Review.
-
Ensemble Calculation for Intrinsically Disordered Proteins Using NMR Parameters.Adv Exp Med Biol. 2015;870:123-47. doi: 10.1007/978-3-319-20164-1_4. Adv Exp Med Biol. 2015. PMID: 26387101 Review.
-
Analytical Description of NMR Relaxation Highlights Correlated Dynamics in Intrinsically Disordered Proteins.Angew Chem Int Ed Engl. 2017 Nov 6;56(45):14020-14024. doi: 10.1002/anie.201706740. Epub 2017 Sep 14. Angew Chem Int Ed Engl. 2017. PMID: 28834051
-
Describing intrinsically disordered proteins at atomic resolution by NMR.Curr Opin Struct Biol. 2013 Jun;23(3):426-35. doi: 10.1016/j.sbi.2013.02.007. Epub 2013 Mar 29. Curr Opin Struct Biol. 2013. PMID: 23545493 Review.
-
Characterization of intrinsically disordered proteins and their dynamic complexes: From in vitro to cell-like environments.Prog Nucl Magn Reson Spectrosc. 2018 Dec;109:79-100. doi: 10.1016/j.pnmrs.2018.07.001. Epub 2018 Jul 31. Prog Nucl Magn Reson Spectrosc. 2018. PMID: 30527137 Review.
Cited by
-
15N-Detected TROSY NMR experiments to study large disordered proteins in high-field magnets.Chem Commun (Camb). 2022 Aug 23;58(68):9512-9515. doi: 10.1039/d2cc02005j. Chem Commun (Camb). 2022. PMID: 35920752 Free PMC article.
-
Nanosecond chain dynamics of single-stranded nucleic acids.Nat Commun. 2024 Jul 17;15(1):6010. doi: 10.1038/s41467-024-50092-8. Nat Commun. 2024. PMID: 39019880 Free PMC article.
-
Structural studies of protein-nucleic acid complexes: A brief overview of the selected techniques.Comput Struct Biotechnol J. 2023 Apr 29;21:2858-2872. doi: 10.1016/j.csbj.2023.04.028. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 37216015 Free PMC article. Review.
-
Predicting the sequence-dependent backbone dynamics of intrinsically disordered proteins.Elife. 2024 Oct 30;12:RP88958. doi: 10.7554/eLife.88958. Elife. 2024. PMID: 39475380 Free PMC article.
-
Data-Driven Models for Predicting Intrinsically Disordered Protein Polymer Physics Directly from Composition or Sequence.Mol Syst Des Eng. 2023 Sep 1;8(9):1146-1155. doi: 10.1039/D3ME00053B. Epub 2023 Jun 6. Mol Syst Des Eng. 2023. PMID: 38222029 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
