

Structural Models for Roseolovirus U20 And U21: Non-Classical MHC-I Like Proteins From HHV-6A, HHV-6B, and HHV-7
- PMID: 35444636
- PMCID: PMC9013968
- DOI: 10.3389/fimmu.2022.864898
Structural Models for Roseolovirus U20 And U21: Non-Classical MHC-I Like Proteins From HHV-6A, HHV-6B, and HHV-7
Erratum in
-
Corrigendum: Structural Models for Roseolovirus U20 And U21: Non-Classical MHC-I Like Proteins From HHV-6A, HHV-6B, and HHV-7.Front Immunol. 2022 May 23;13:936968. doi: 10.3389/fimmu.2022.936968. eCollection 2022. Front Immunol. 2022. PMID: 35677042 Free PMC article.
Abstract
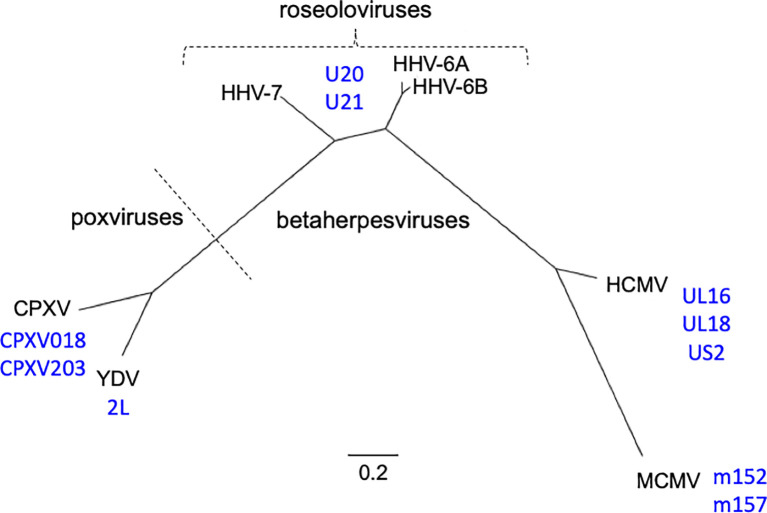
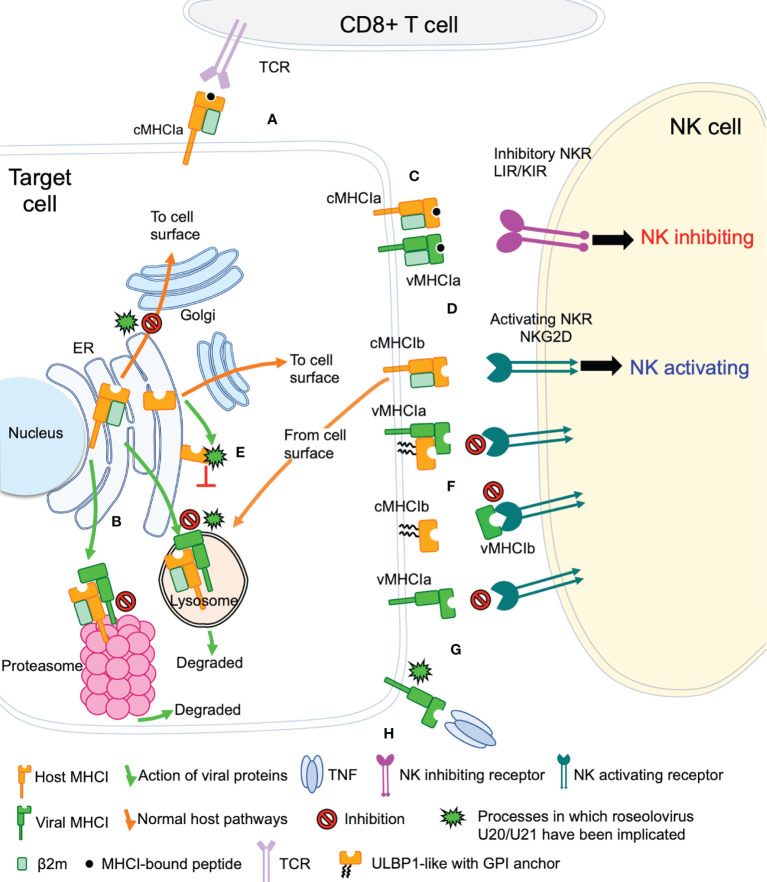
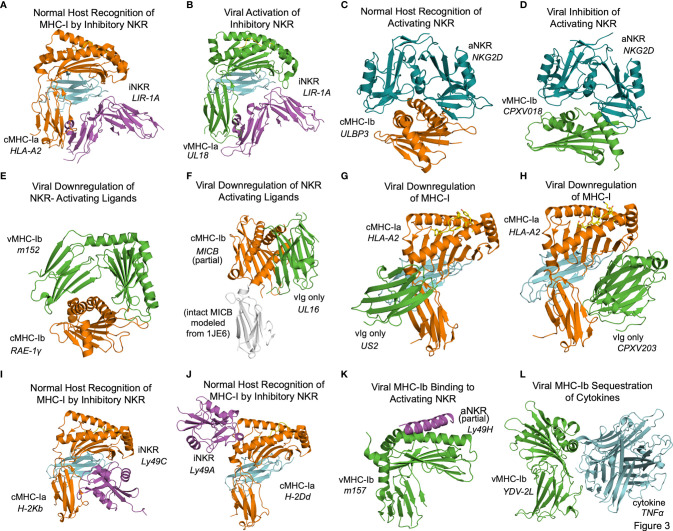
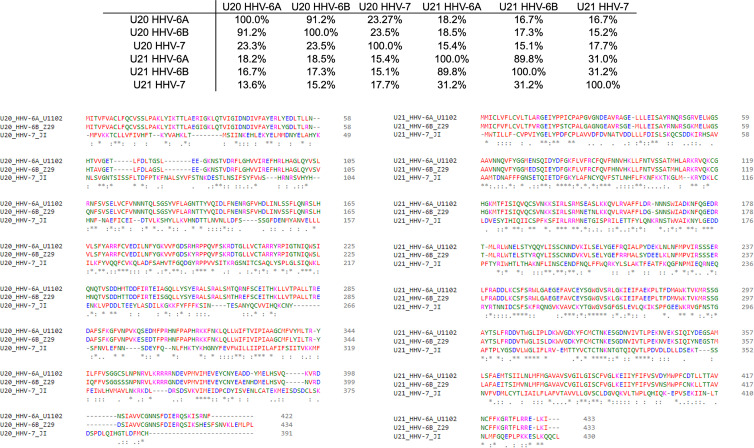
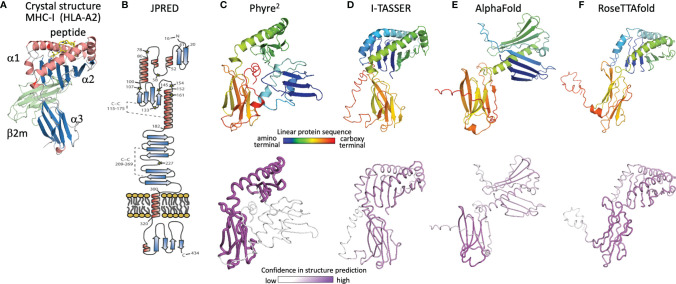
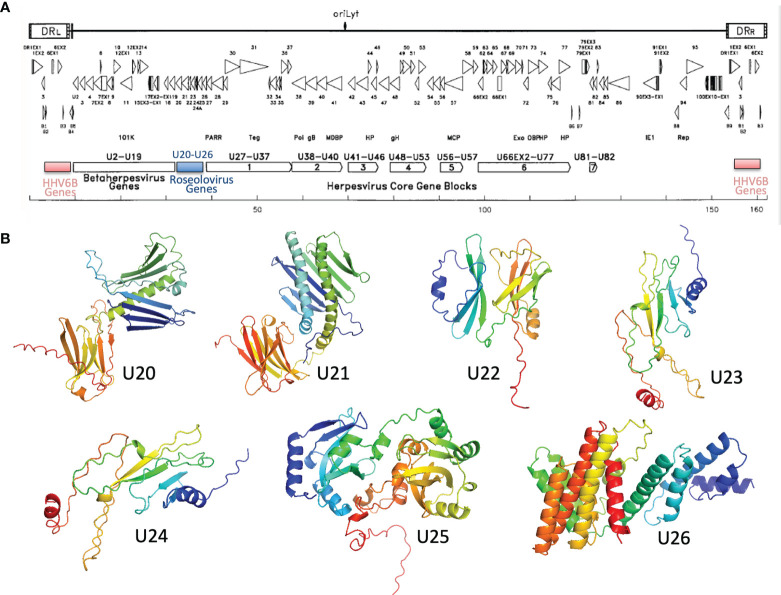
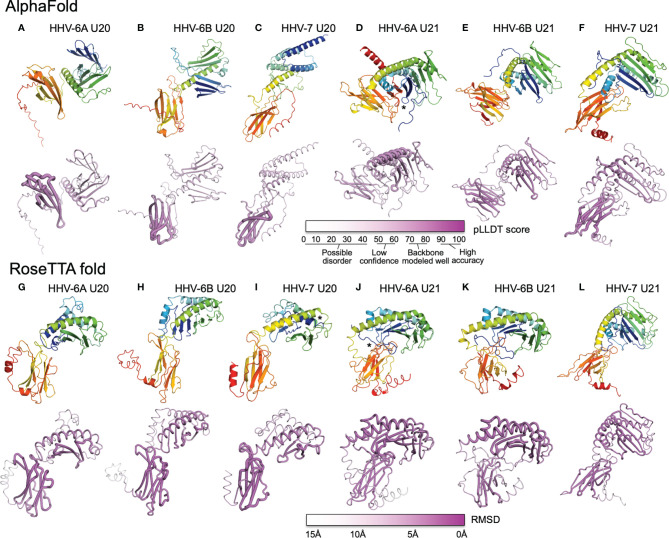
Human roseolovirus U20 and U21 are type I membrane glycoproteins that have been implicated in immune evasion by interfering with recognition of classical and non-classical MHC proteins. U20 and U21 are predicted to be type I glycoproteins with extracytosolic immunoglobulin-like domains, but detailed structural information is lacking. AlphaFold and RoseTTAfold are next generation machine-learning-based prediction engines that recently have revolutionized the field of computational three-dimensional protein structure prediction. Here, we review the structural biology of viral immunoevasins and the current status of computational structure prediction algorithms. We use these computational tools to generate structural models for U20 and U21 proteins, which are predicted to adopt MHC-Ia-like folds with closed MHC platforms and immunoglobulin-like domains. We evaluate these structural models and place them within current understanding of the structural basis for viral immune evasion of T cell and natural killer cell recognition.
Keywords: MHC1b; human herpesvirus; immune recognition; immunoevasion; machine learning; major histocompatibility protein; natural killer cell ligand; structure prediction.
Copyright © 2022 Weaver, Arya, Schneider, Hudson and Stern.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
The HHV-6B U20 glycoprotein binds ULBP1, masking it from recognition by NKG2D and interfering with natural killer cell activation.Front Immunol. 2024 Jun 17;15:1363156. doi: 10.3389/fimmu.2024.1363156. eCollection 2024. Front Immunol. 2024. PMID: 38953028 Free PMC article.
-
Corrigendum: Structural Models for Roseolovirus U20 And U21: Non-Classical MHC-I Like Proteins From HHV-6A, HHV-6B, and HHV-7.Front Immunol. 2022 May 23;13:936968. doi: 10.3389/fimmu.2022.936968. eCollection 2022. Front Immunol. 2022. PMID: 35677042 Free PMC article.
-
The HHV-6A Proteins U20 and U21 Target NKG2D Ligands to Escape Immune Recognition.Front Immunol. 2021 Oct 15;12:714799. doi: 10.3389/fimmu.2021.714799. eCollection 2021. Front Immunol. 2021. PMID: 34721381 Free PMC article.
-
Glycoproteins of HHV-6A and HHV-6B.Adv Exp Med Biol. 2018;1045:145-165. doi: 10.1007/978-981-10-7230-7_8. Adv Exp Med Biol. 2018. PMID: 29896667 Review.
-
Roseolovirus molecular biology: recent advances.Curr Opin Virol. 2014 Dec;9:170-7. doi: 10.1016/j.coviro.2014.10.004. Epub 2014 Nov 27. Curr Opin Virol. 2014. PMID: 25437229 Free PMC article. Review.
Cited by
-
Immunopeptidome profiling of human coronavirus OC43-infected cells identifies CD4 T cell epitopes specific to seasonal coronaviruses or cross-reactive with SARS-CoV-2.bioRxiv [Preprint]. 2022 Dec 1:2022.12.01.518643. doi: 10.1101/2022.12.01.518643. bioRxiv. 2022. Update in: PLoS Pathog. 2023 Jul 27;19(7):e1011032. doi: 10.1371/journal.ppat.1011032. PMID: 36482973 Free PMC article. Updated. Preprint.
-
Characterization of the HHV-6B U20 Immunoevasin.J Virol. 2023 Feb 28;97(2):e0189022. doi: 10.1128/jvi.01890-22. Epub 2023 Jan 23. J Virol. 2023. PMID: 36688652 Free PMC article.
-
Immunopeptidome profiling of human coronavirus OC43-infected cells identifies CD4 T-cell epitopes specific to seasonal coronaviruses or cross-reactive with SARS-CoV-2.PLoS Pathog. 2023 Jul 27;19(7):e1011032. doi: 10.1371/journal.ppat.1011032. eCollection 2023 Jul. PLoS Pathog. 2023. PMID: 37498934 Free PMC article.
-
The HHV-6B U20 glycoprotein binds ULBP1, masking it from recognition by NKG2D and interfering with natural killer cell activation.Front Immunol. 2024 Jun 17;15:1363156. doi: 10.3389/fimmu.2024.1363156. eCollection 2024. Front Immunol. 2024. PMID: 38953028 Free PMC article.
-
Human herpesvirus 7 meningitis in an adolescent with normal immune function: A case report.World J Clin Cases. 2024 Jun 26;12(18):3636-3643. doi: 10.12998/wjcc.v12.i18.3636. World J Clin Cases. 2024. PMID: 38983391 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials