

Inter-paralog amino acid inversion events in large phylogenies of duplicated proteins
- PMID: 35377869
- PMCID: PMC9009777
- DOI: 10.1371/journal.pcbi.1010016
Inter-paralog amino acid inversion events in large phylogenies of duplicated proteins
Abstract
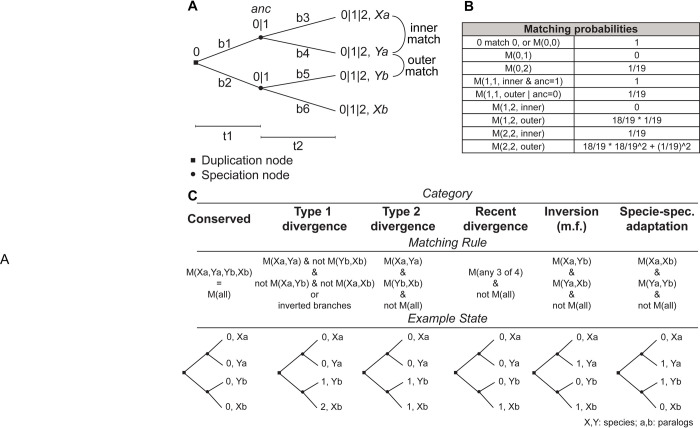
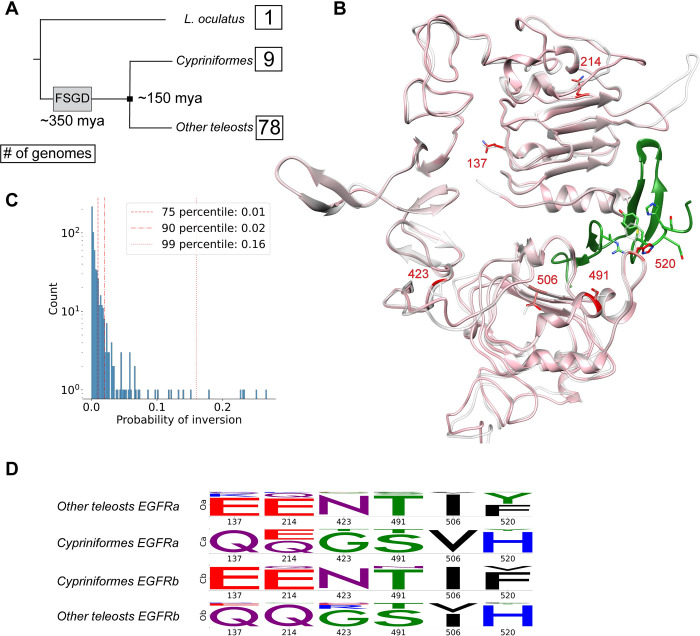
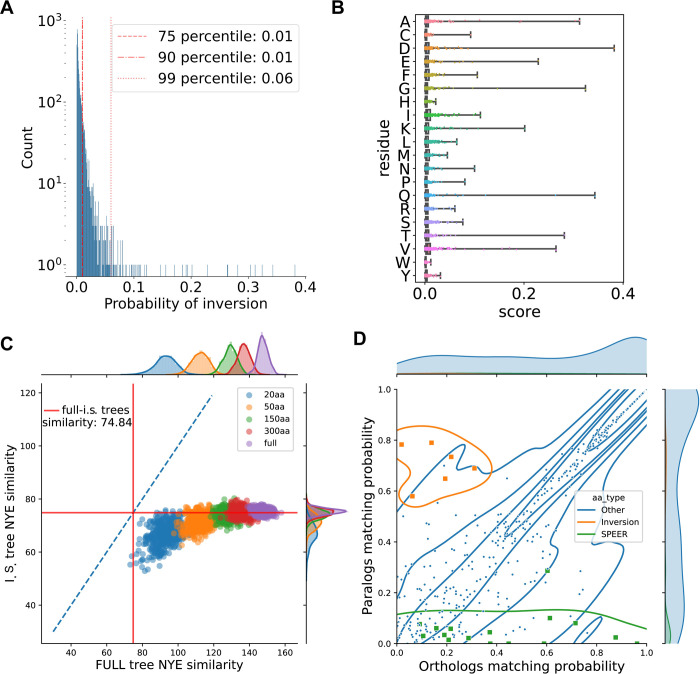
Connecting protein sequence to function is becoming increasingly relevant since high-throughput sequencing studies accumulate large amounts of genomic data. In order to go beyond the existing database annotation, it is fundamental to understand the mechanisms underlying functional inheritance and divergence. If the homology relationship between proteins is known, can we determine whether the function diverged? In this work, we analyze different possibilities of protein sequence evolution after gene duplication and identify "inter-paralog inversions", i.e., sites where the relationship between the ancestry and the functional signal is decoupled. The amino acids in these sites are masked from being recognized by other prediction tools. Still, they play a role in functional divergence and could indicate a shift in protein function. We develop a method to specifically recognize inter-paralog amino acid inversions in a phylogeny and test it on real and simulated datasets. In a dataset built from the Epidermal Growth Factor Receptor (EGFR) sequences found in 88 fish species, we identify 19 amino acid sites that went through inversion after gene duplication, mostly located at the ligand-binding extracellular domain. Our work uncovers an outcome of protein duplications with direct implications in protein functional annotation and sequence evolution. The developed method is optimized to work with large protein datasets and can be readily included in a targeted protein analysis pipeline.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


Similar articles
-
An update of DIVERGE software for functional divergence analysis of protein family.Mol Biol Evol. 2013 Jul;30(7):1713-9. doi: 10.1093/molbev/mst069. Epub 2013 Apr 14. Mol Biol Evol. 2013. PMID: 23589455
-
Dealing with saturation at the amino acid level: a case study based on anciently duplicated zebrafish genes.Gene. 2002 Aug 7;295(2):205-11. doi: 10.1016/s0378-1119(02)00689-3. Gene. 2002. PMID: 12354655
-
Evolutionary patterns of amino acid substitutions in 12 Drosophila genomes.BMC Genomics. 2010 Dec 2;11 Suppl 4(Suppl 4):S10. doi: 10.1186/1471-2164-11-S4-S10. BMC Genomics. 2010. PMID: 21143793 Free PMC article.
-
Evolution of duplications in the transferrin family of proteins.Comp Biochem Physiol B Biochem Mol Biol. 2005 Jan;140(1):11-25. doi: 10.1016/j.cbpc.2004.09.012. Comp Biochem Physiol B Biochem Mol Biol. 2005. PMID: 15621505 Review.
-
Evolution of signal transduction by gene and genome duplication in fish.J Struct Funct Genomics. 2003;3(1-4):139-50. J Struct Funct Genomics. 2003. PMID: 12836693 Review.
Cited by
-
Paralog-based synthetic lethality: rationales and applications.Front Oncol. 2023 Jun 7;13:1168143. doi: 10.3389/fonc.2023.1168143. eCollection 2023. Front Oncol. 2023. PMID: 37350942 Free PMC article. Review.
-
How gene duplication diversifies the landscape of protein oligomeric state and function.Curr Opin Genet Dev. 2022 Oct;76:101966. doi: 10.1016/j.gde.2022.101966. Epub 2022 Aug 22. Curr Opin Genet Dev. 2022. PMID: 36007298 Free PMC article. Review.
References
-
- Rockah-Shmuel L, Tóth-Petróczy Á, Tawfik DS. Systematic Mapping of Protein Mutational Space by Prolonged Drift Reveals the Deleterious Effects of Seemingly Neutral Mutations. PLoS Comput Biol. 2015;11(8):e1004421. Epub 2015/08/15. doi: 10.1371/journal.pcbi.1004421 ; PubMed Central PMCID: PMC4537296. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
