

Multifunctional Lipid Bilayer Nanocarriers for Cancer Immunotherapy in Heterogeneous Tumor Microenvironments, Combining Immunogenic Cell Death Stimuli with Immune Modulatory Drugs
- PMID: 35348320
- PMCID: PMC9519818
- DOI: 10.1021/acsnano.2c01252
Multifunctional Lipid Bilayer Nanocarriers for Cancer Immunotherapy in Heterogeneous Tumor Microenvironments, Combining Immunogenic Cell Death Stimuli with Immune Modulatory Drugs
Abstract
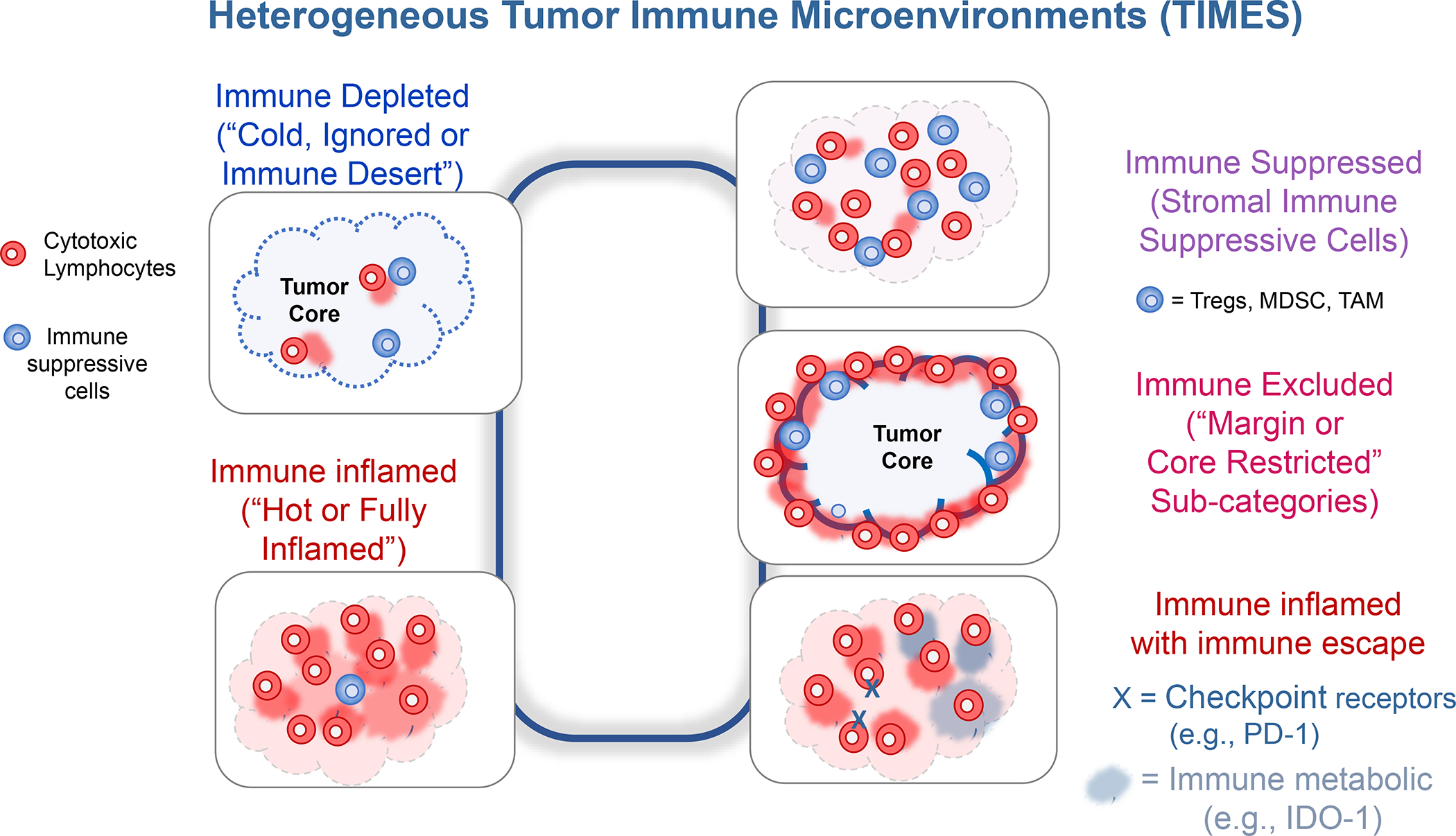
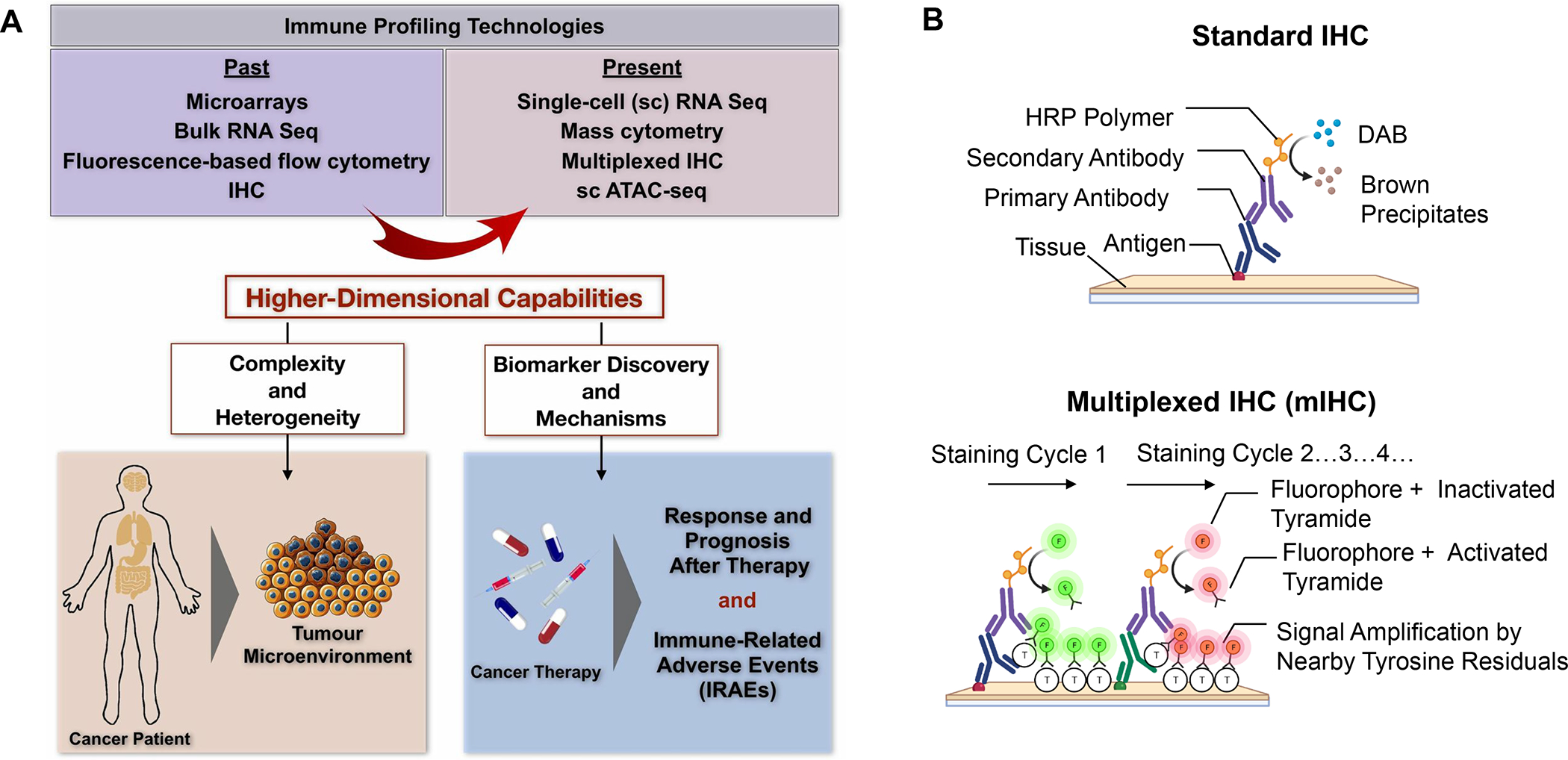
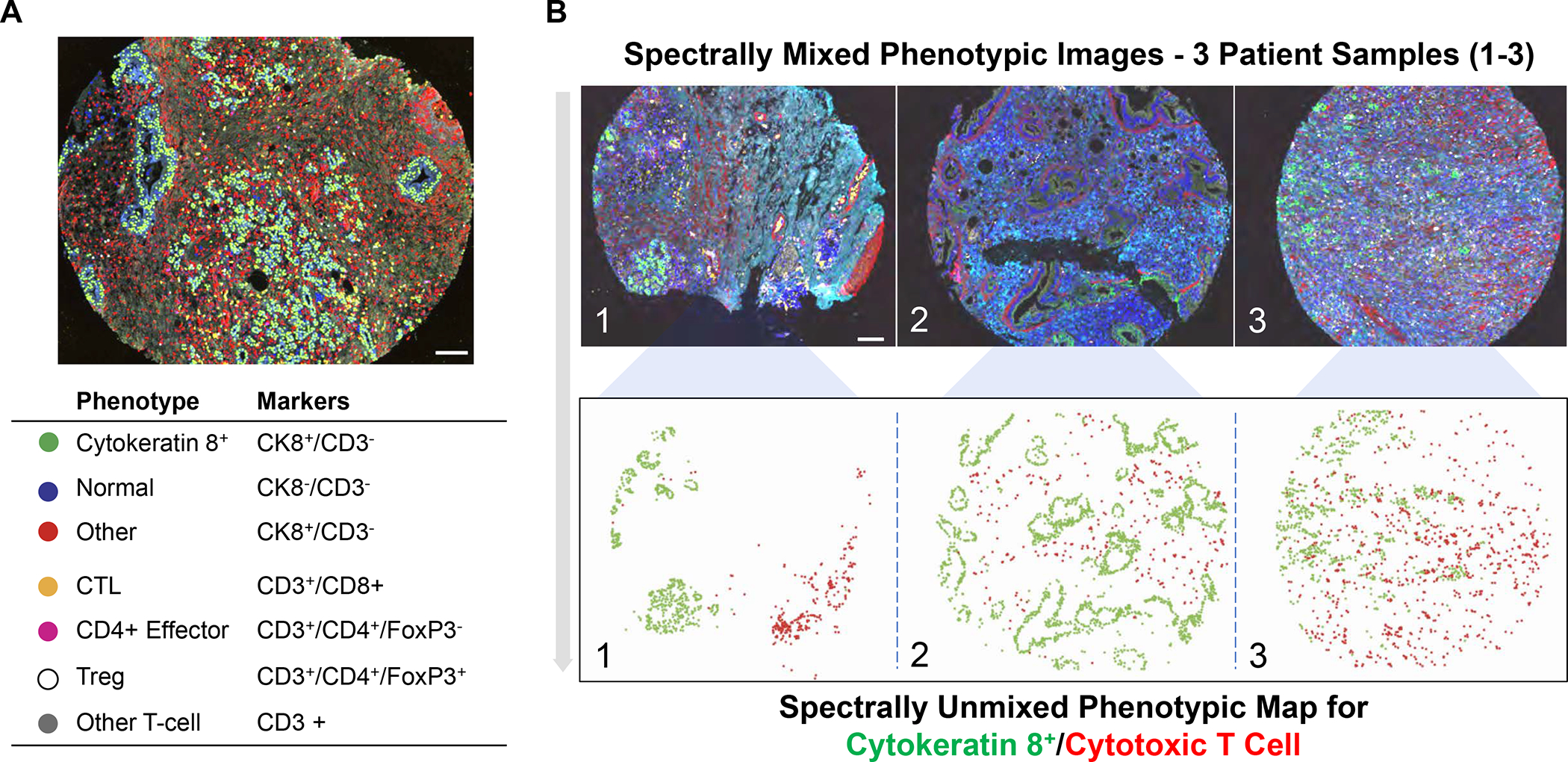
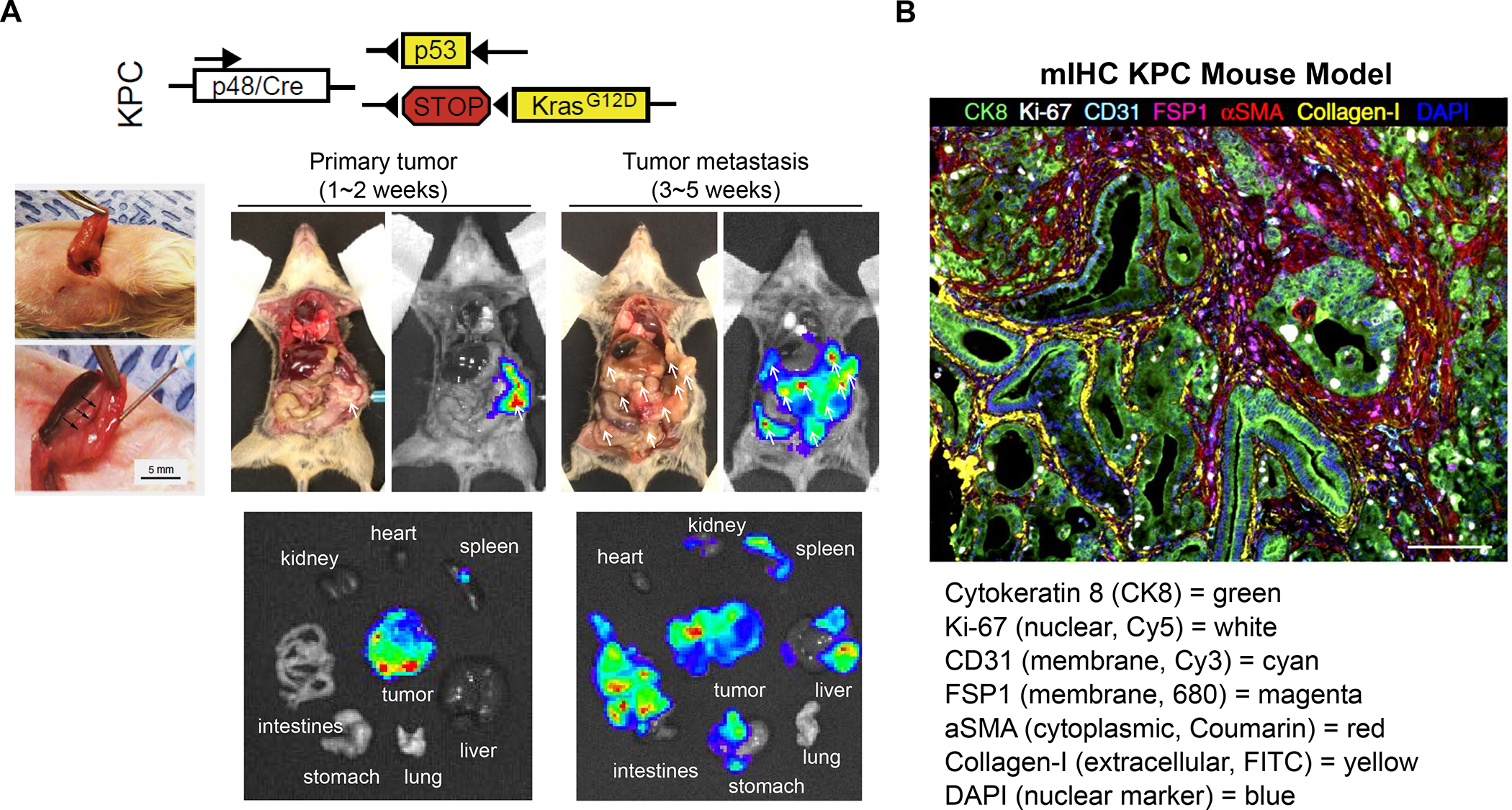
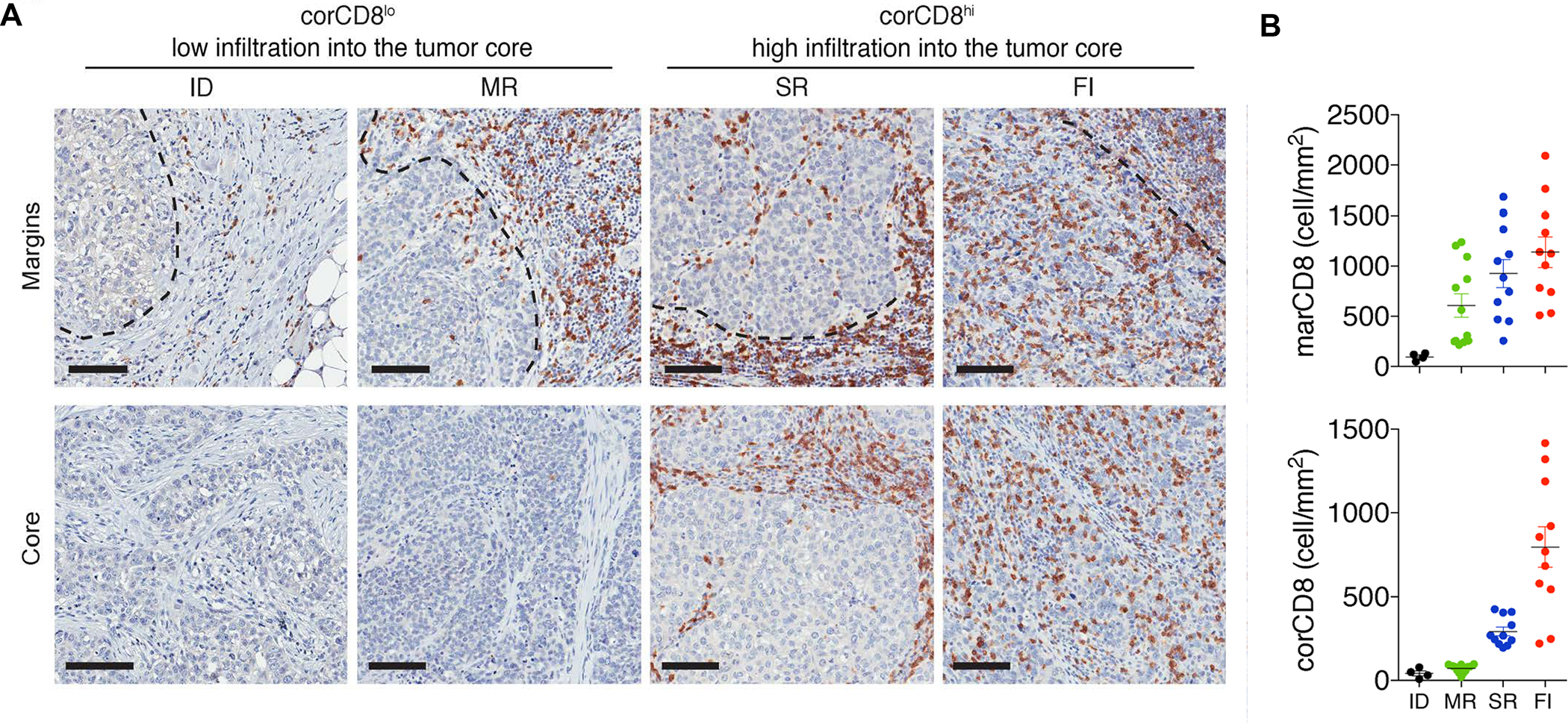
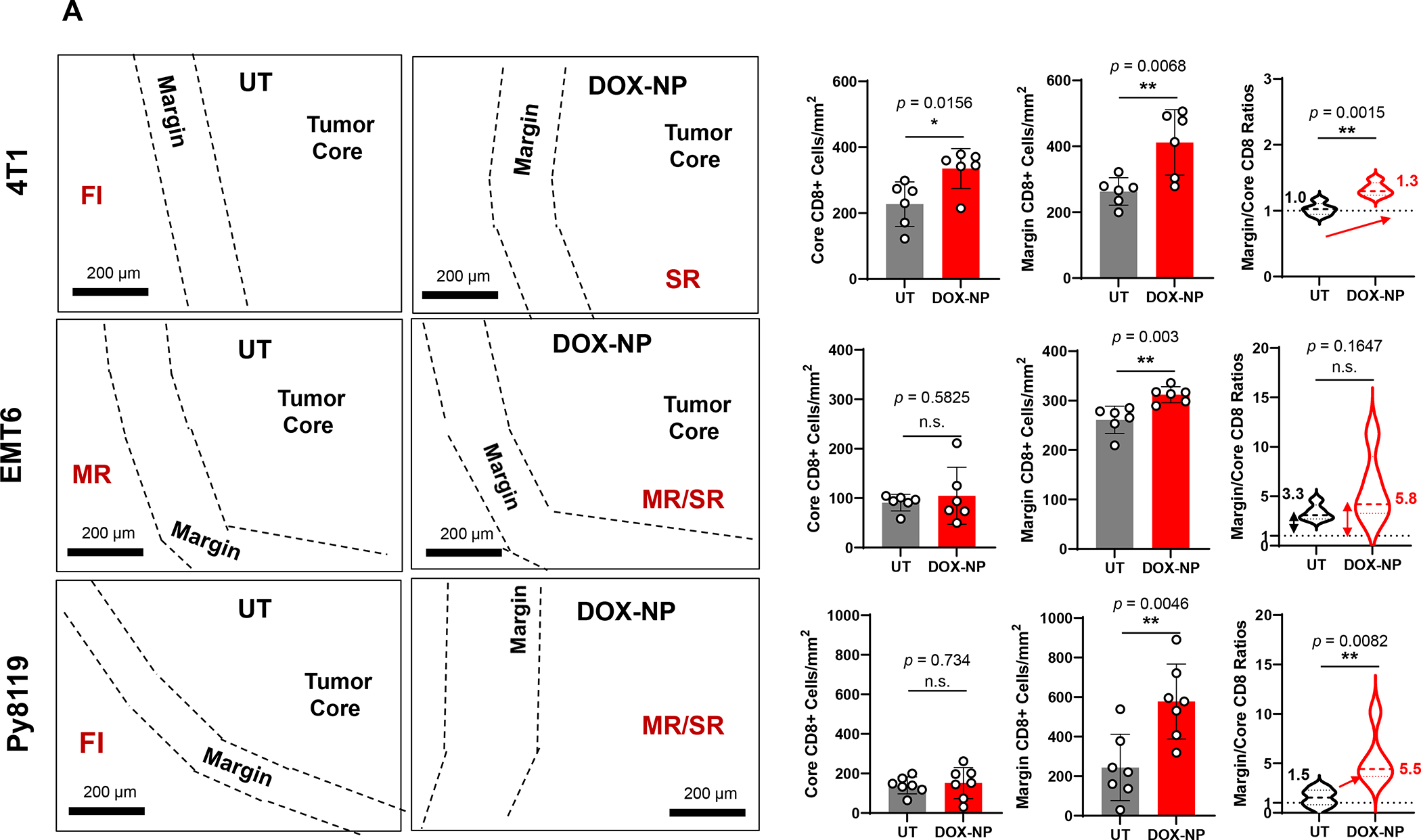
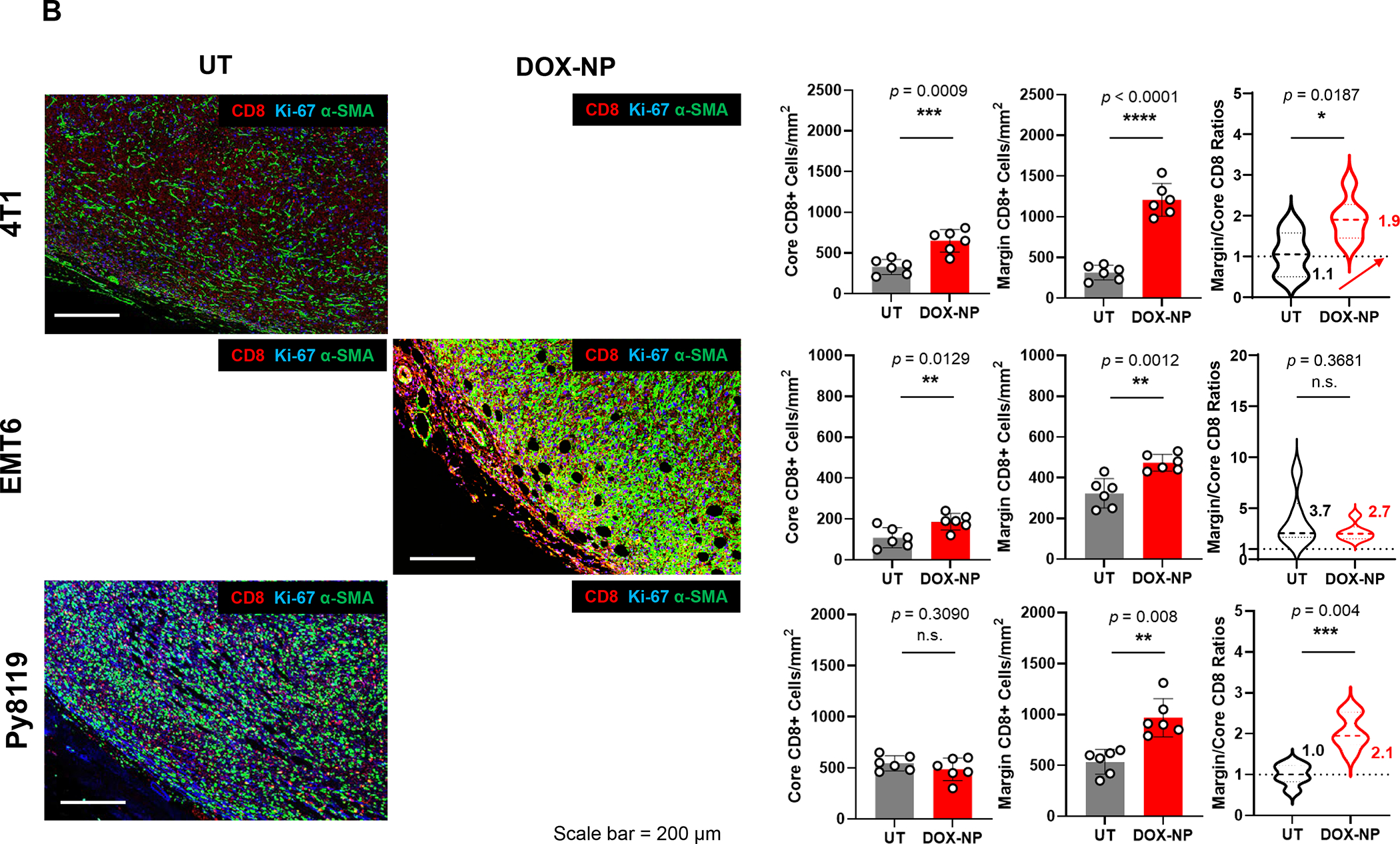
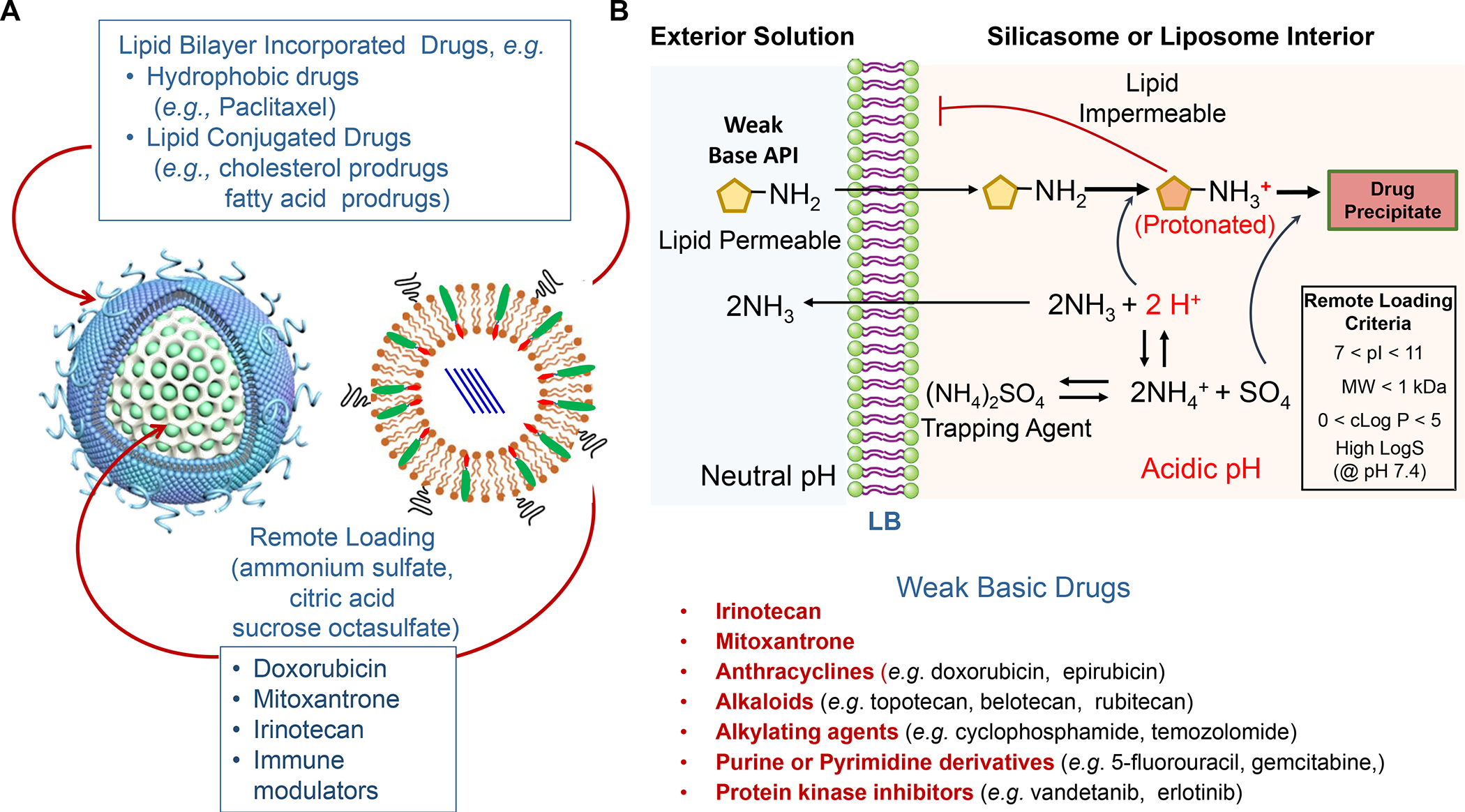
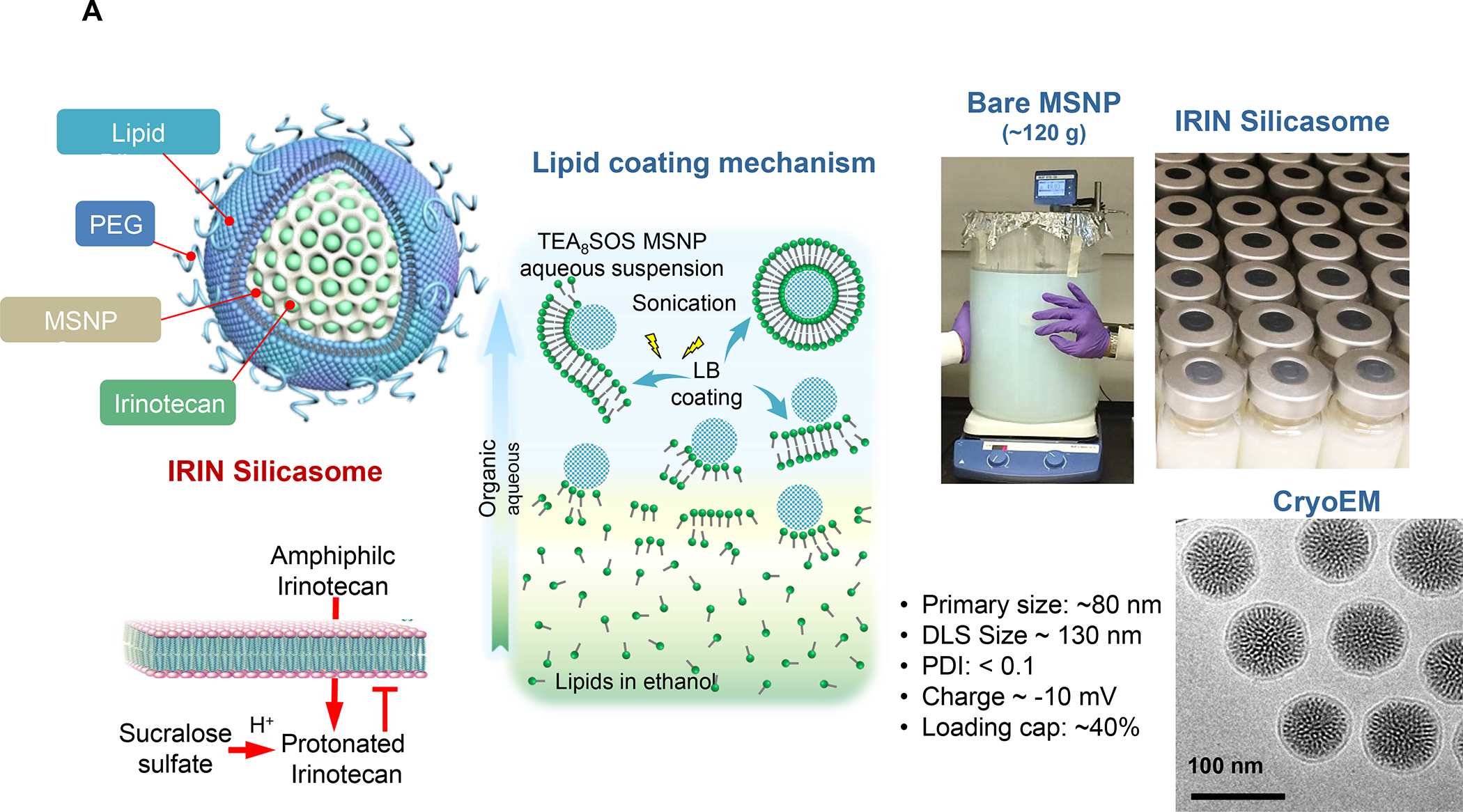
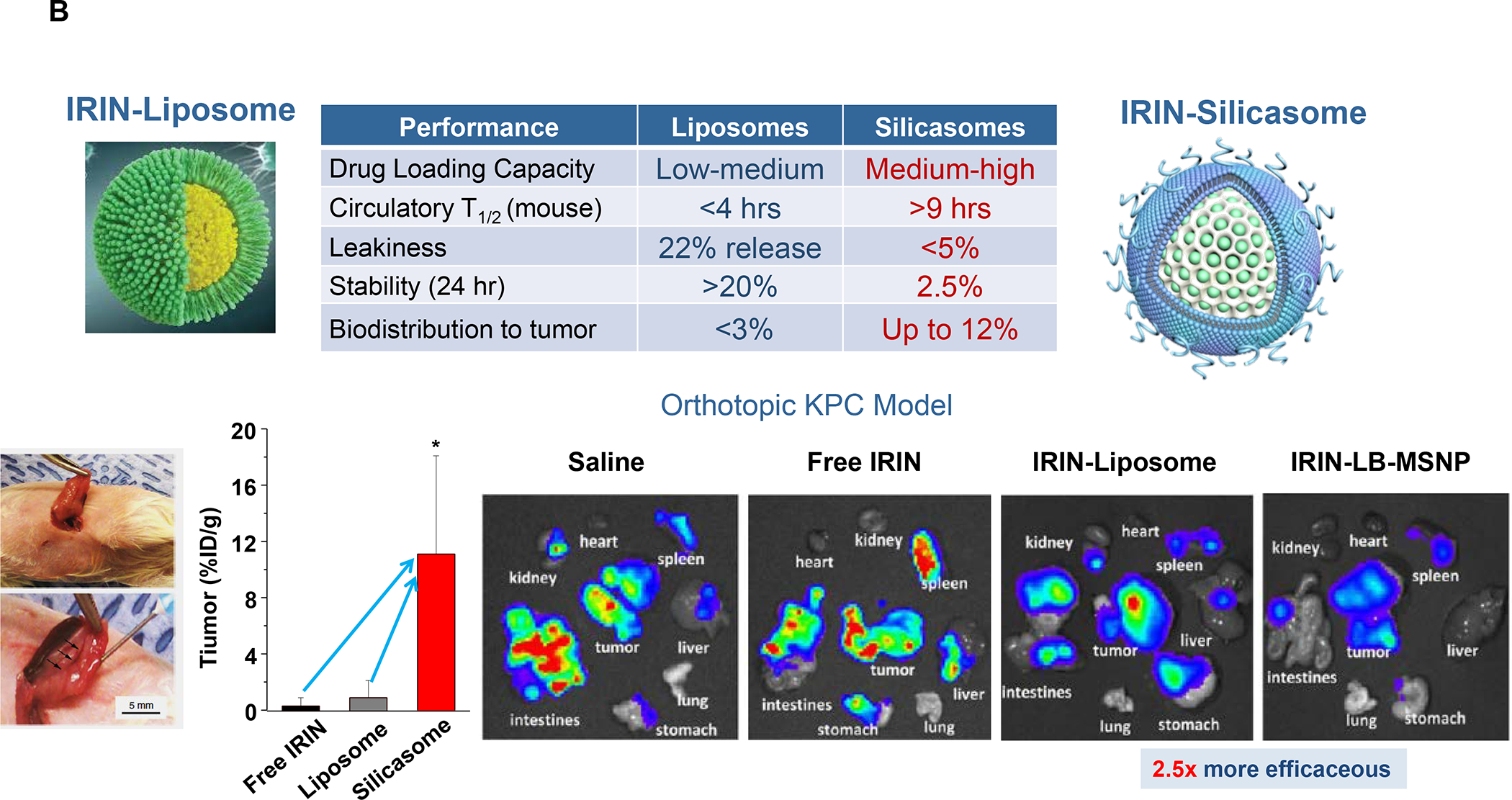
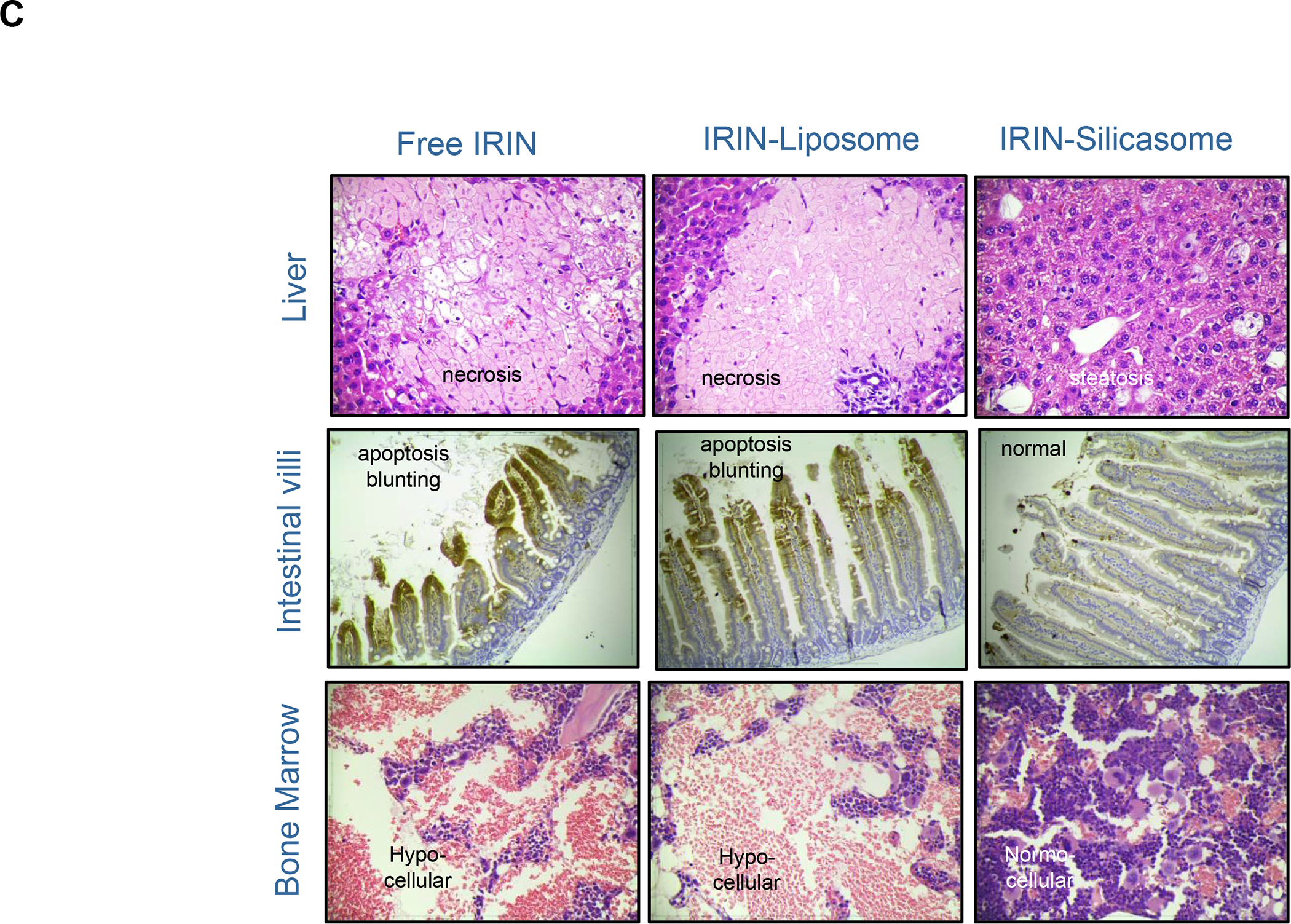
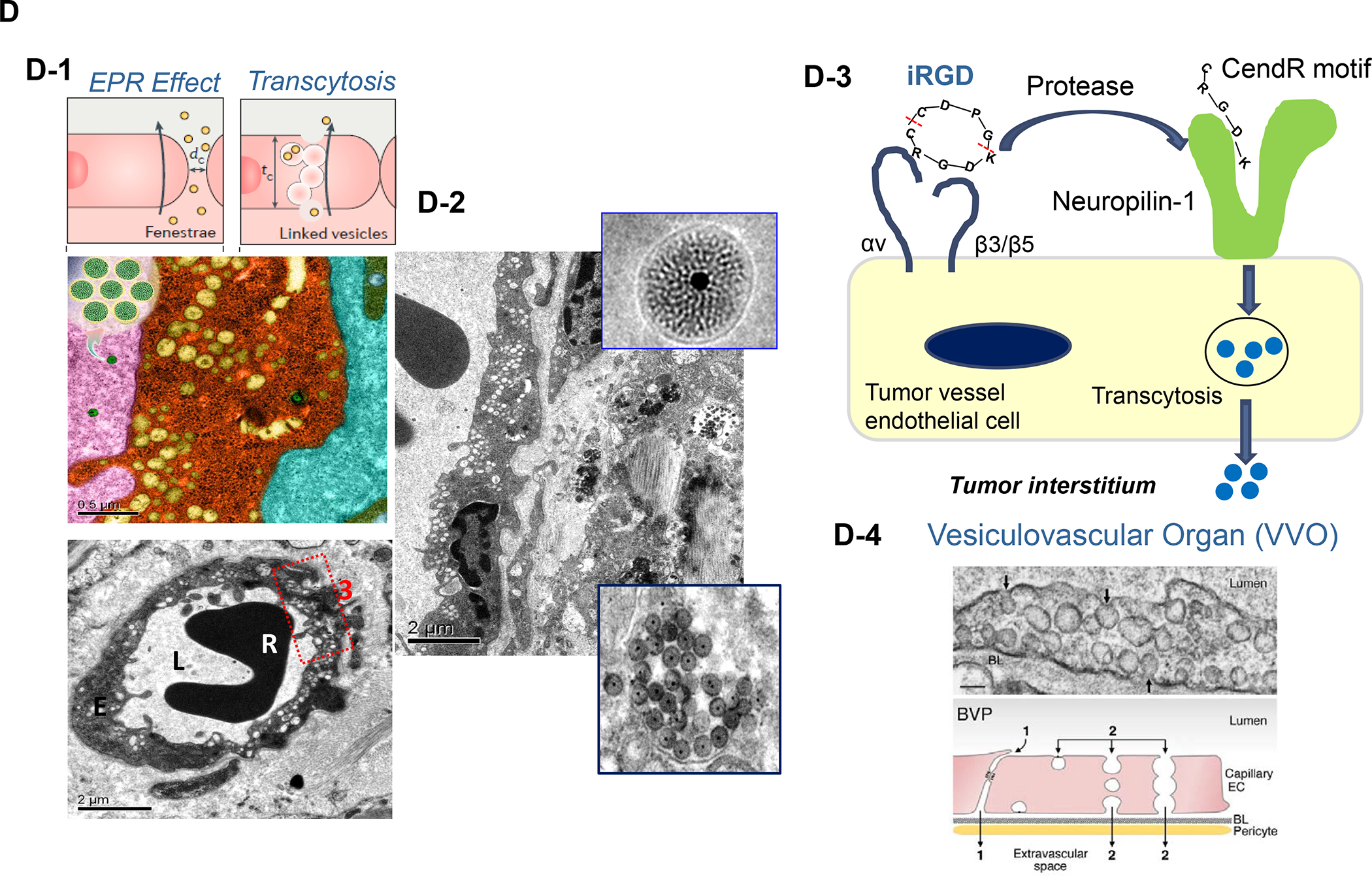
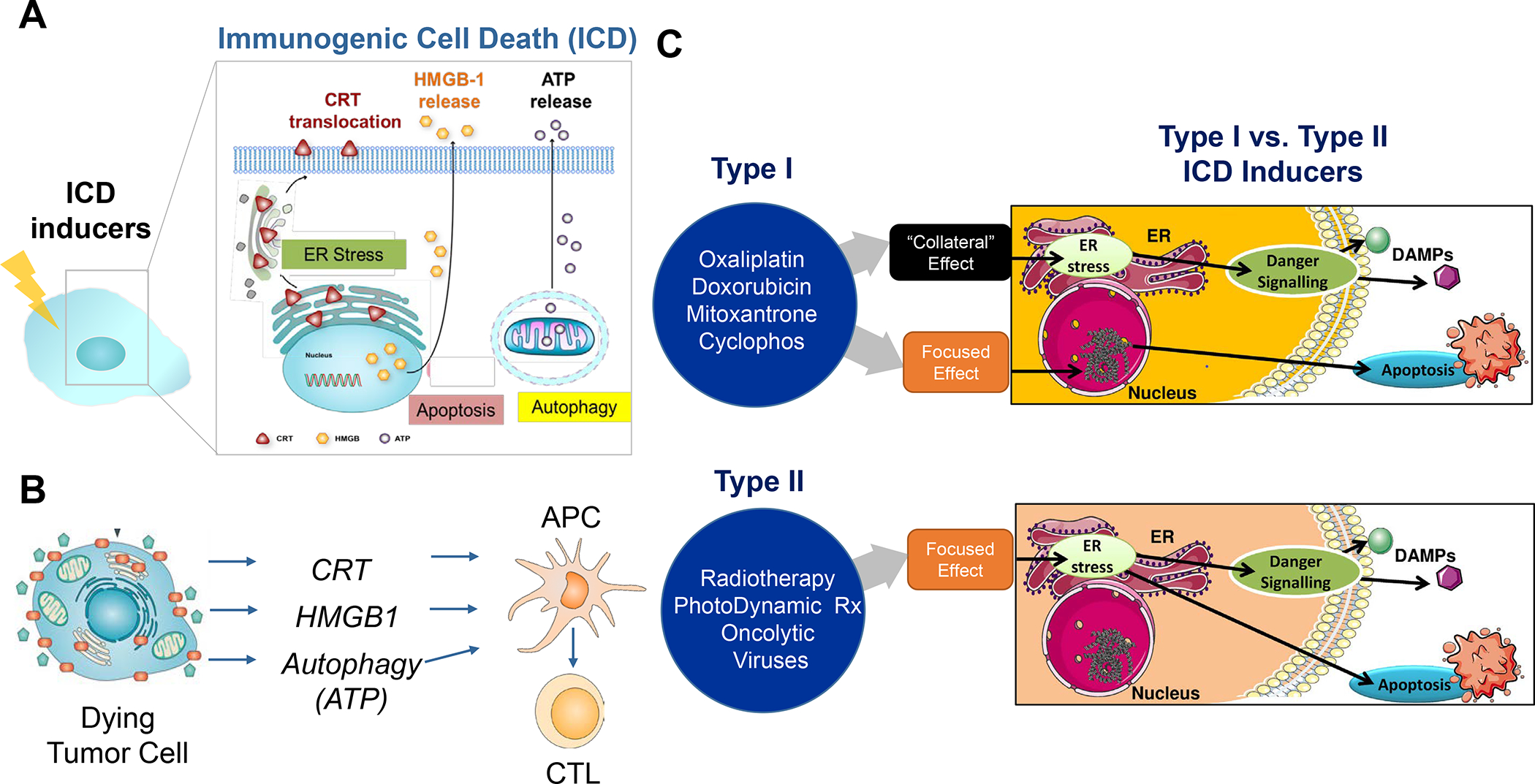
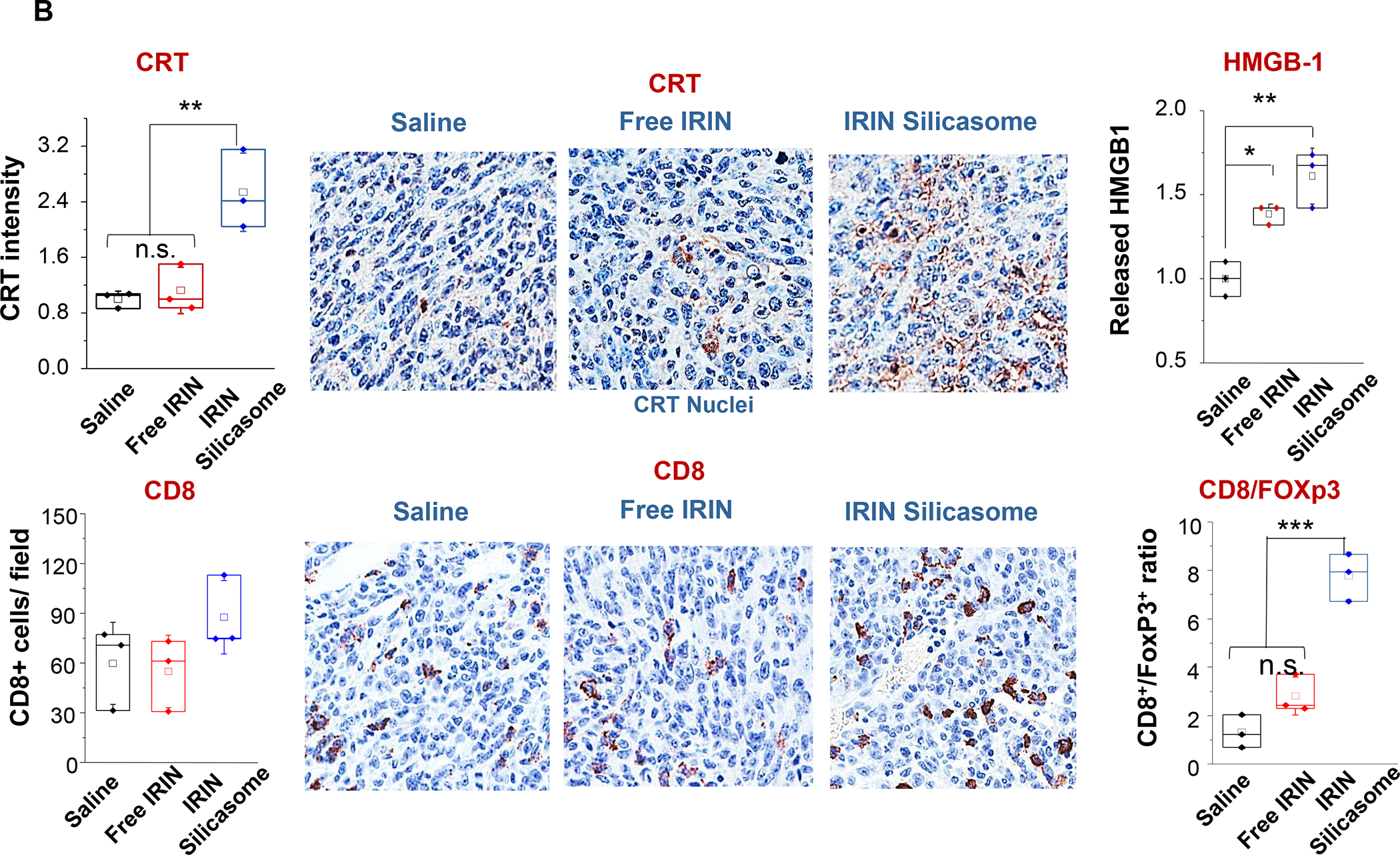
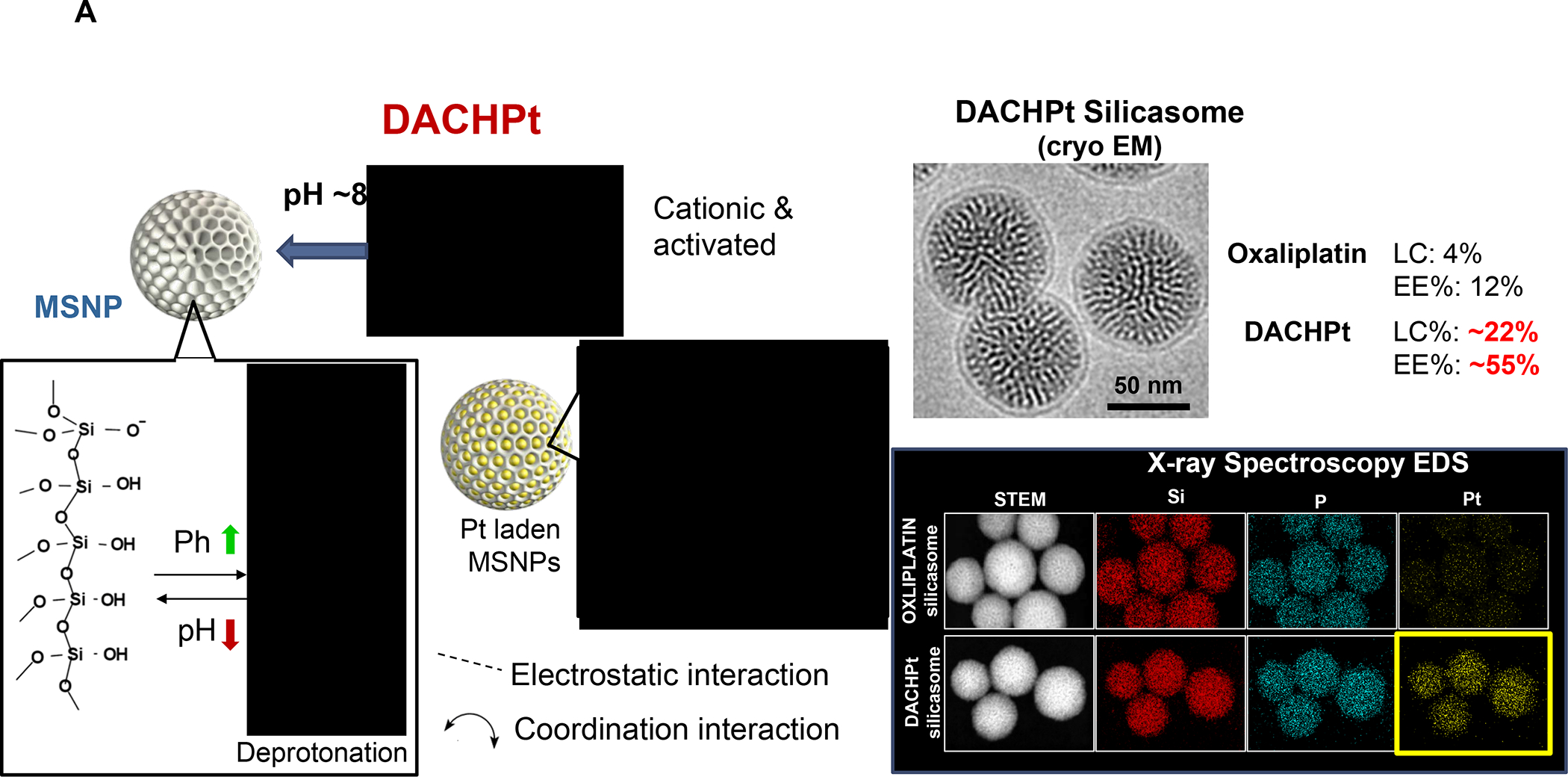
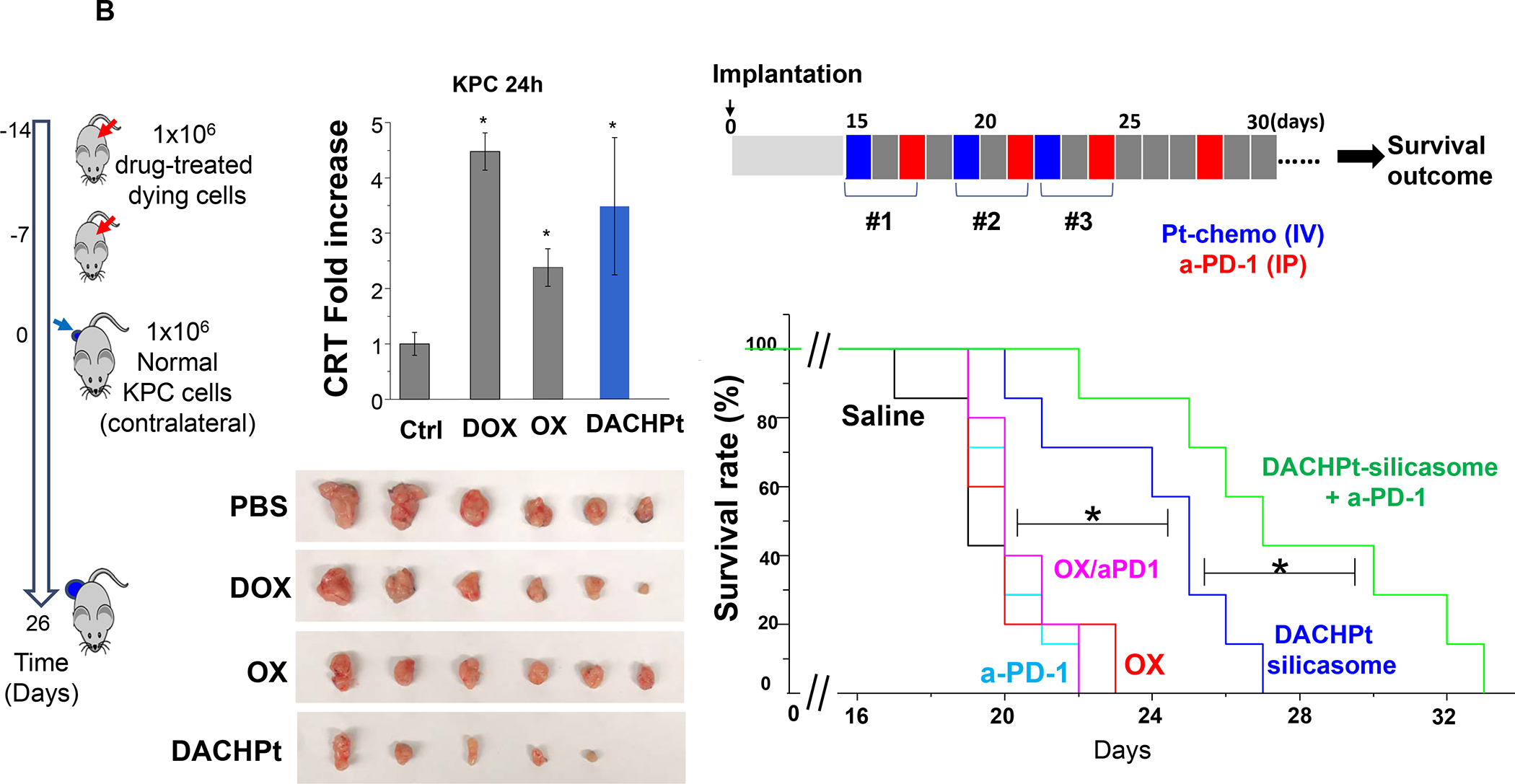
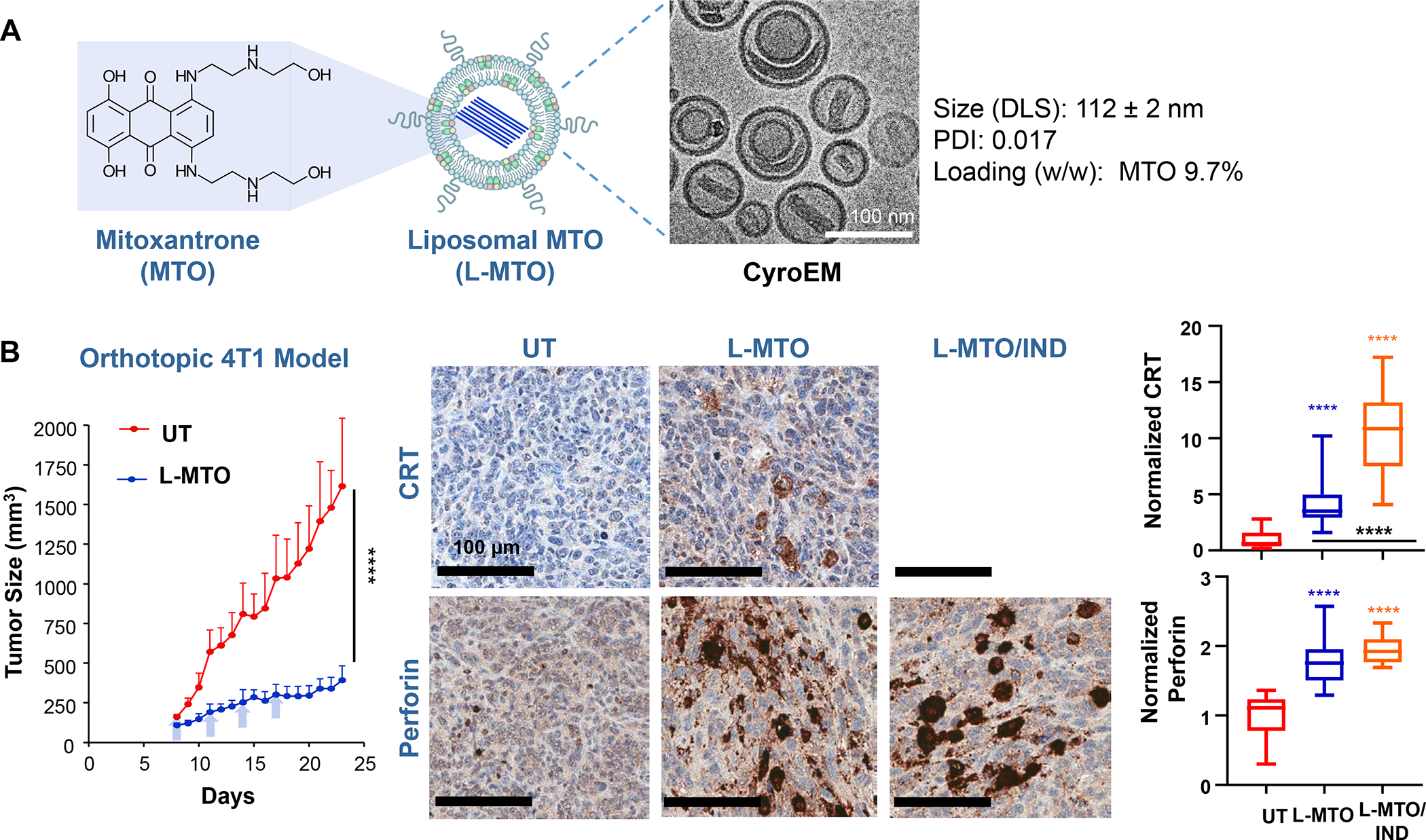
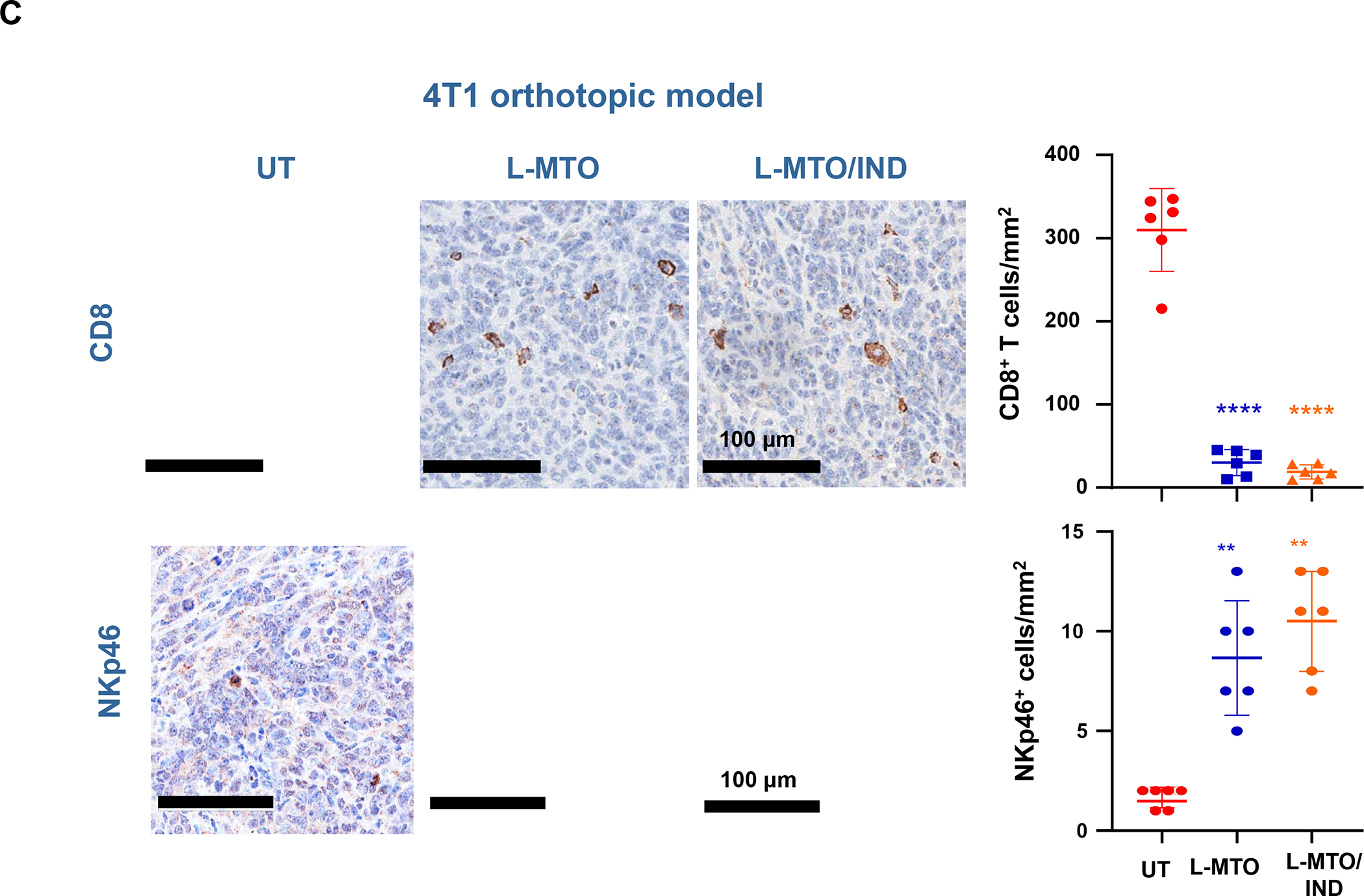
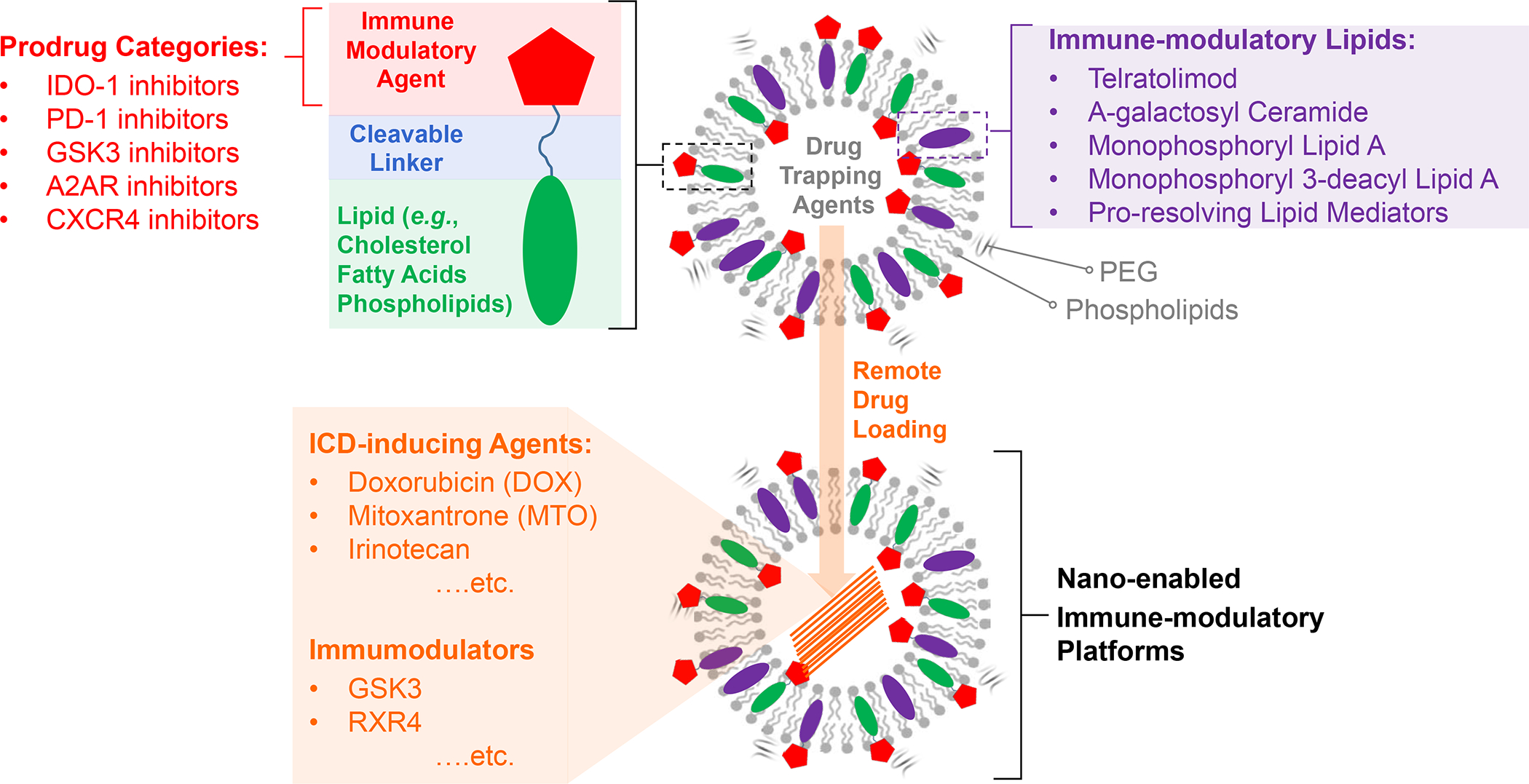
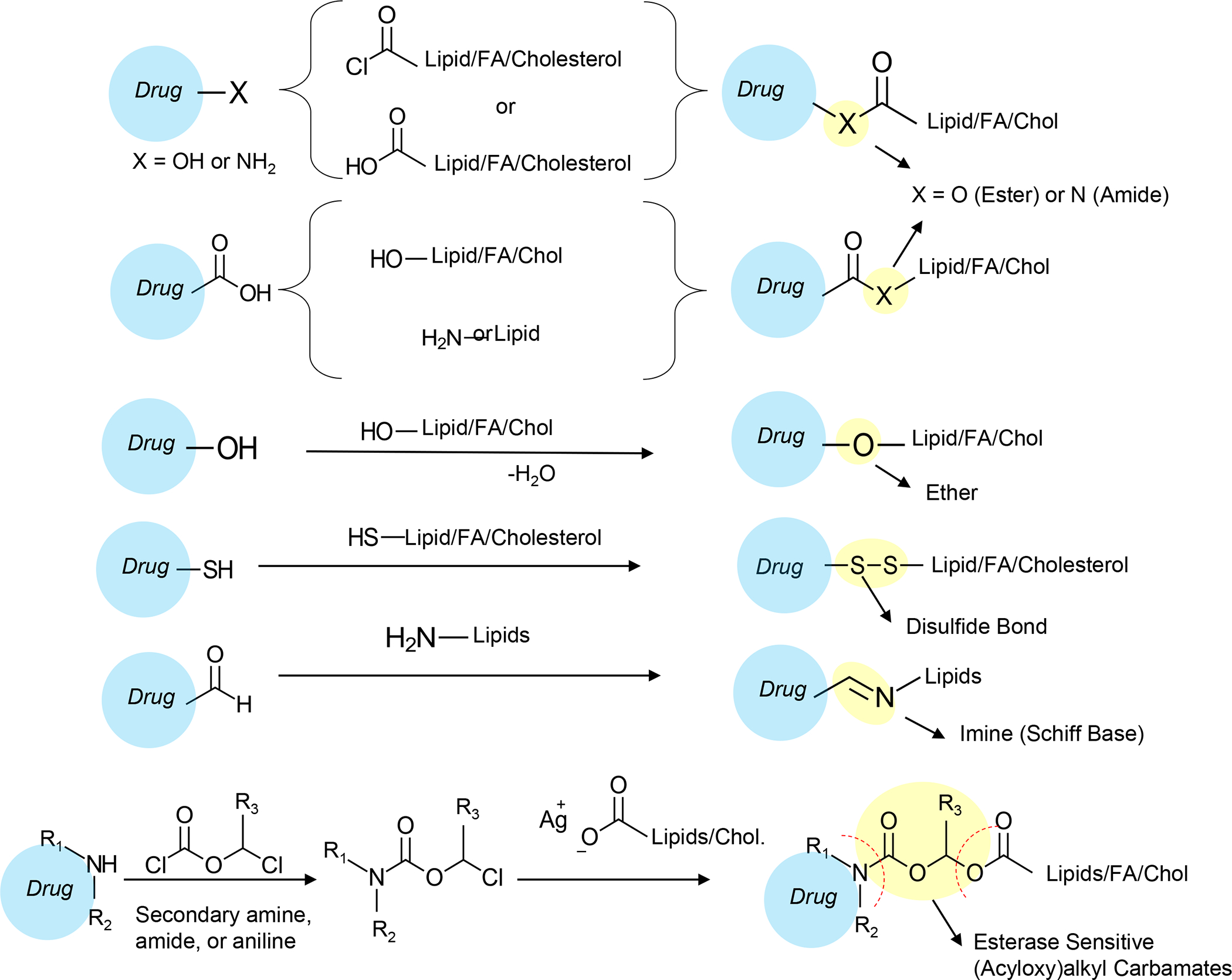
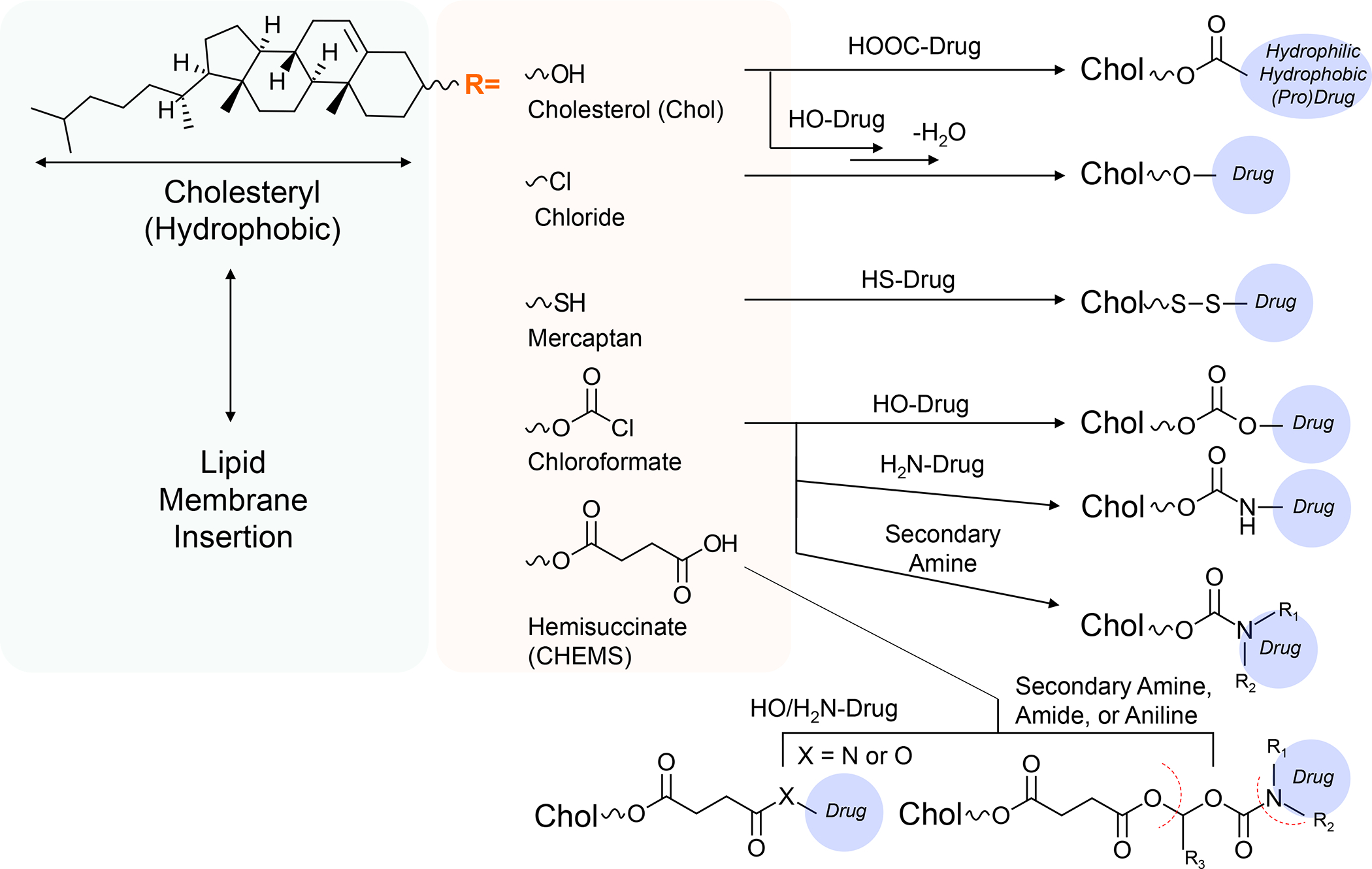
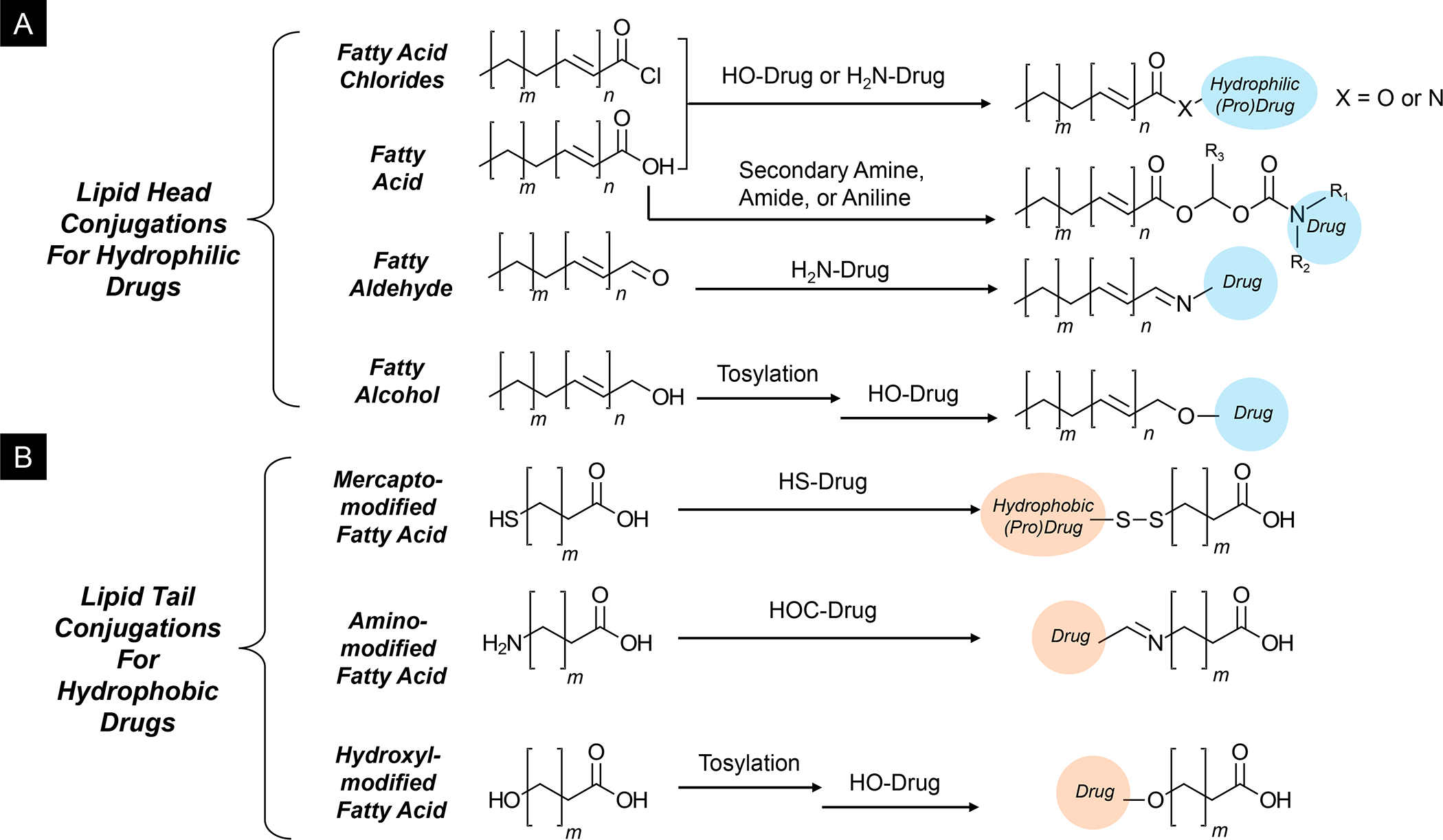
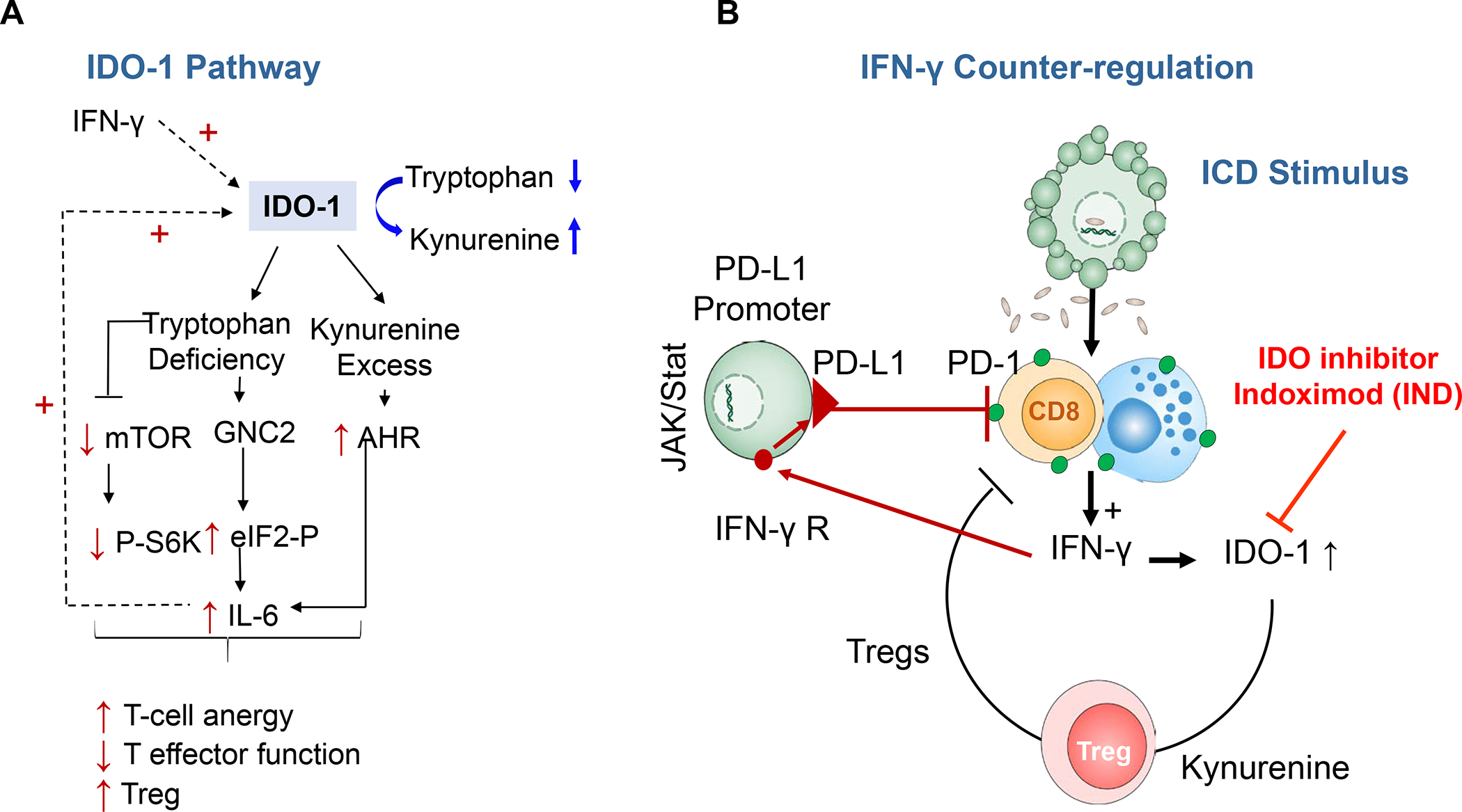
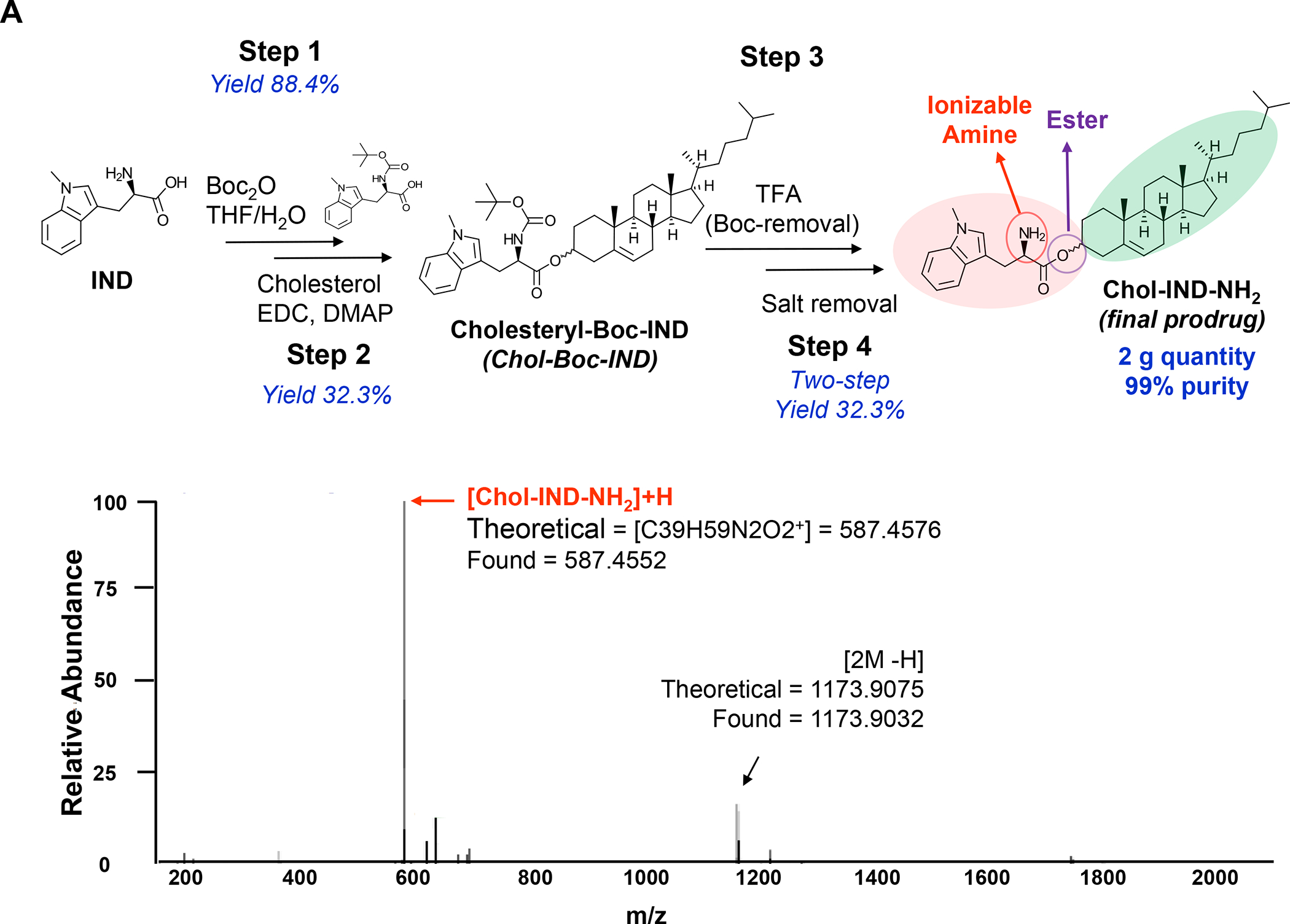
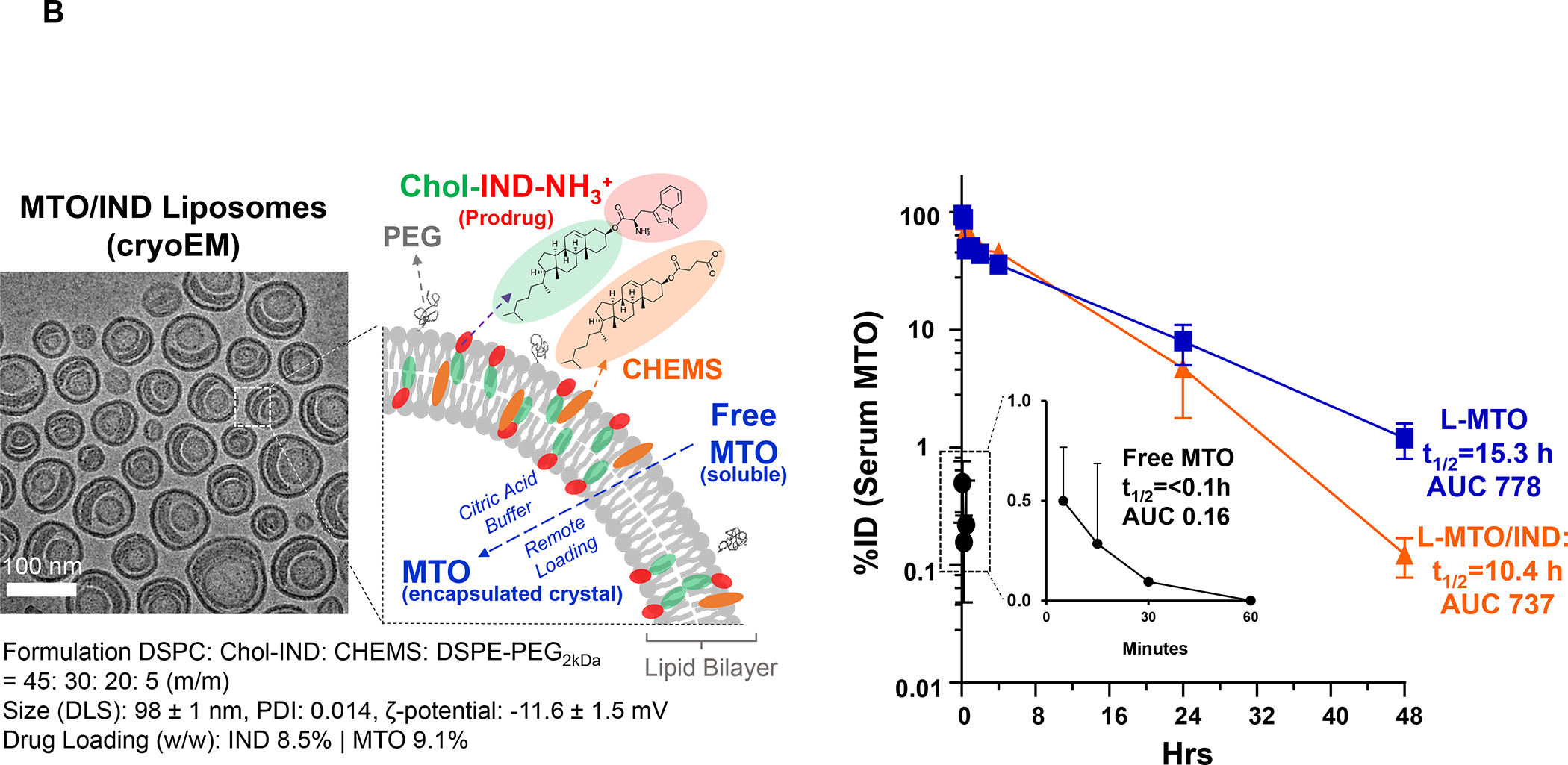
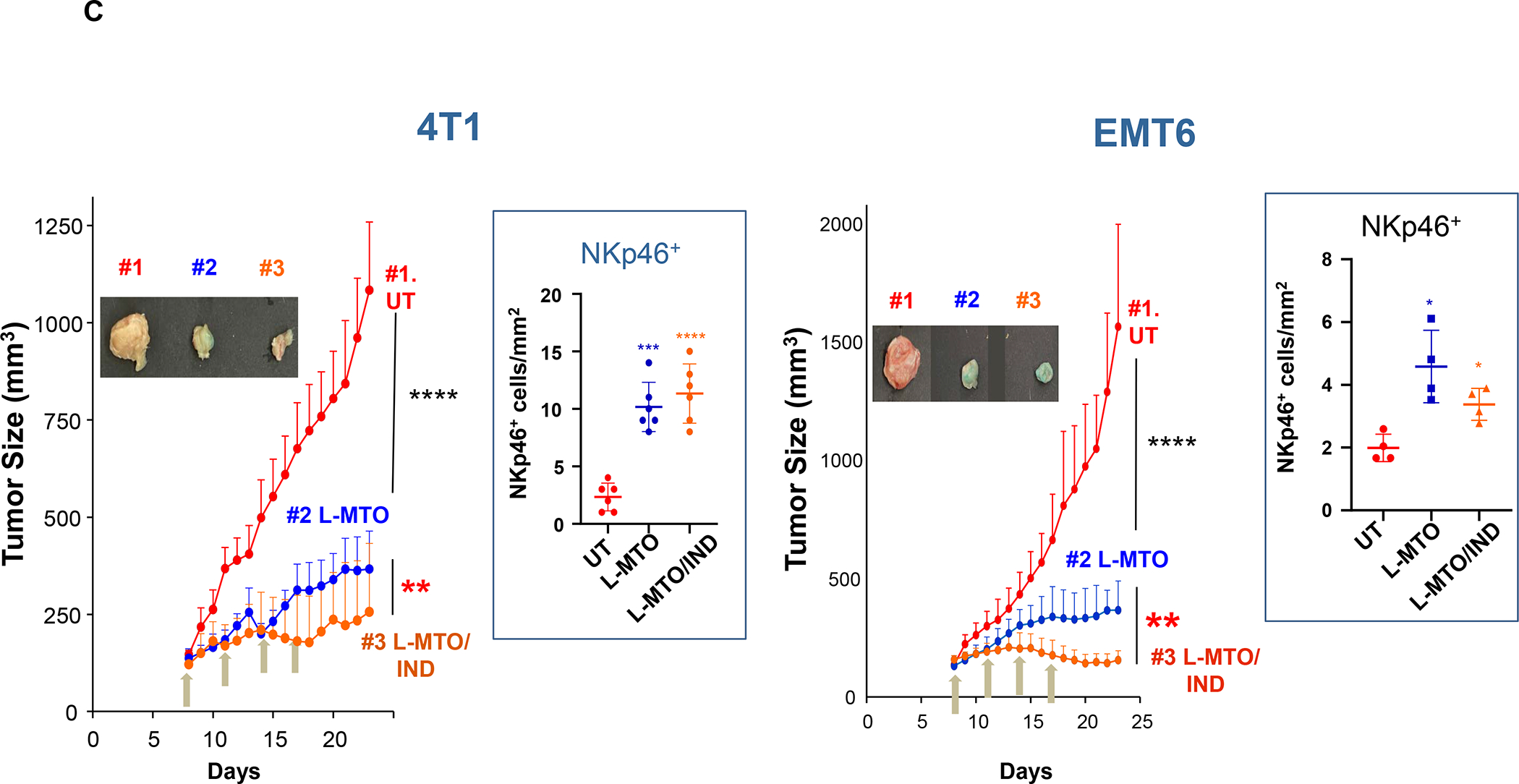
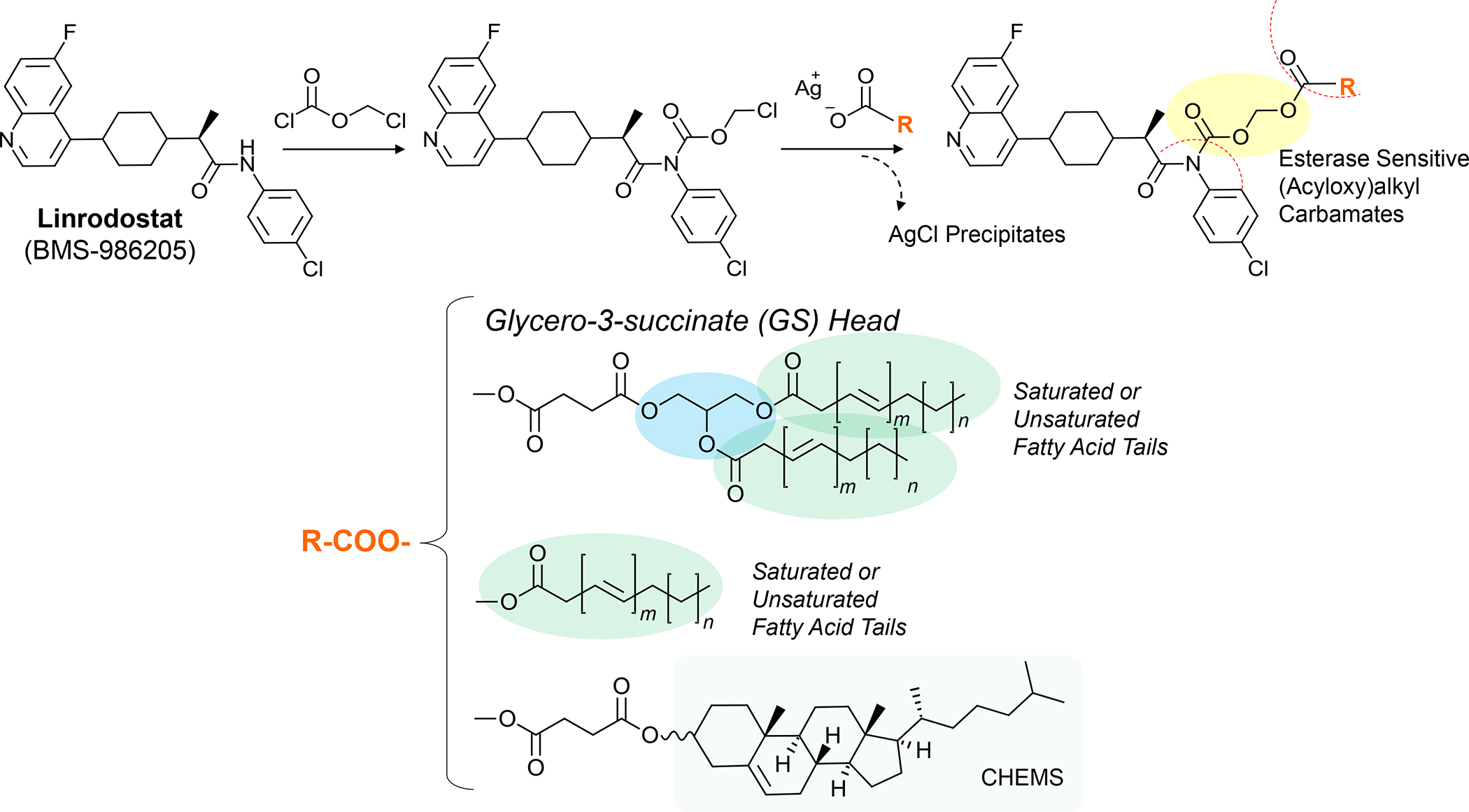
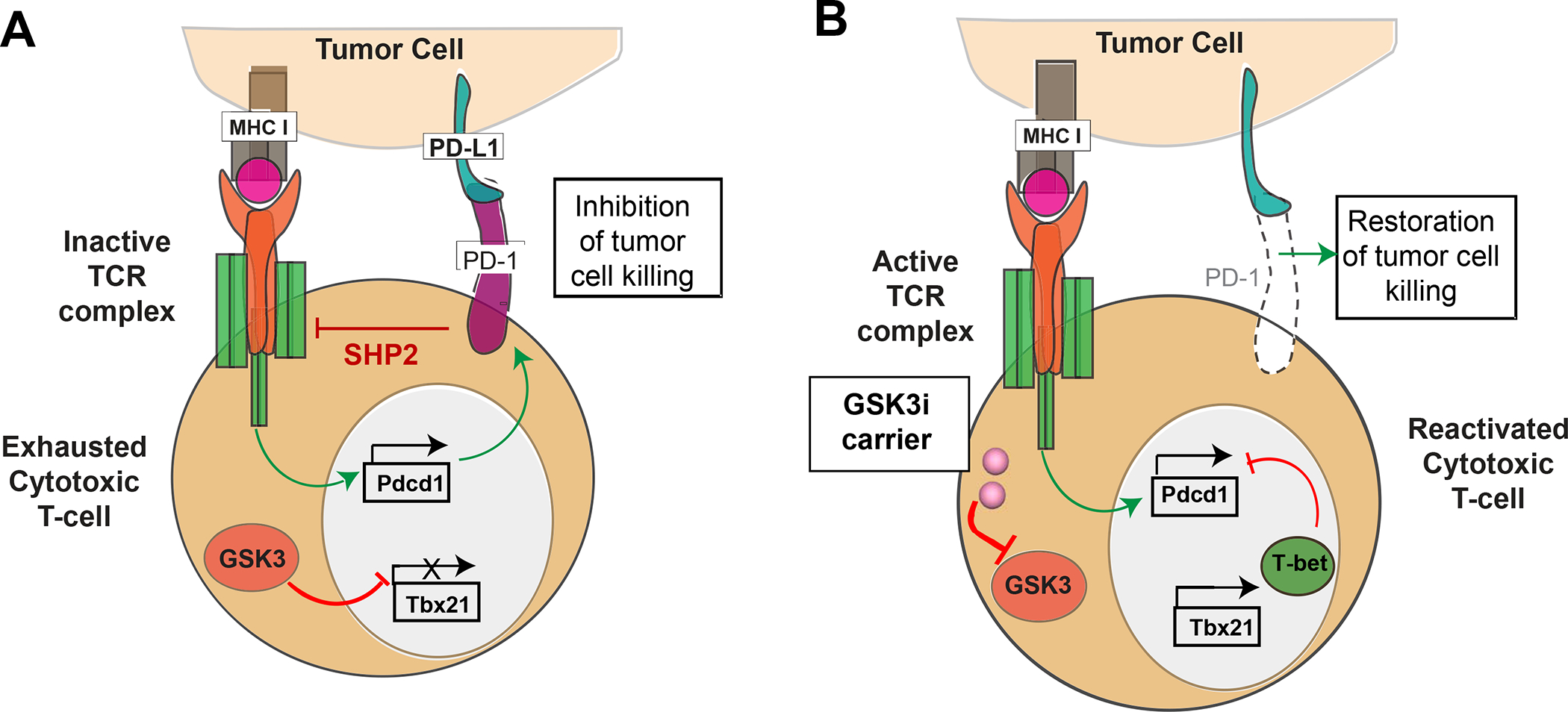
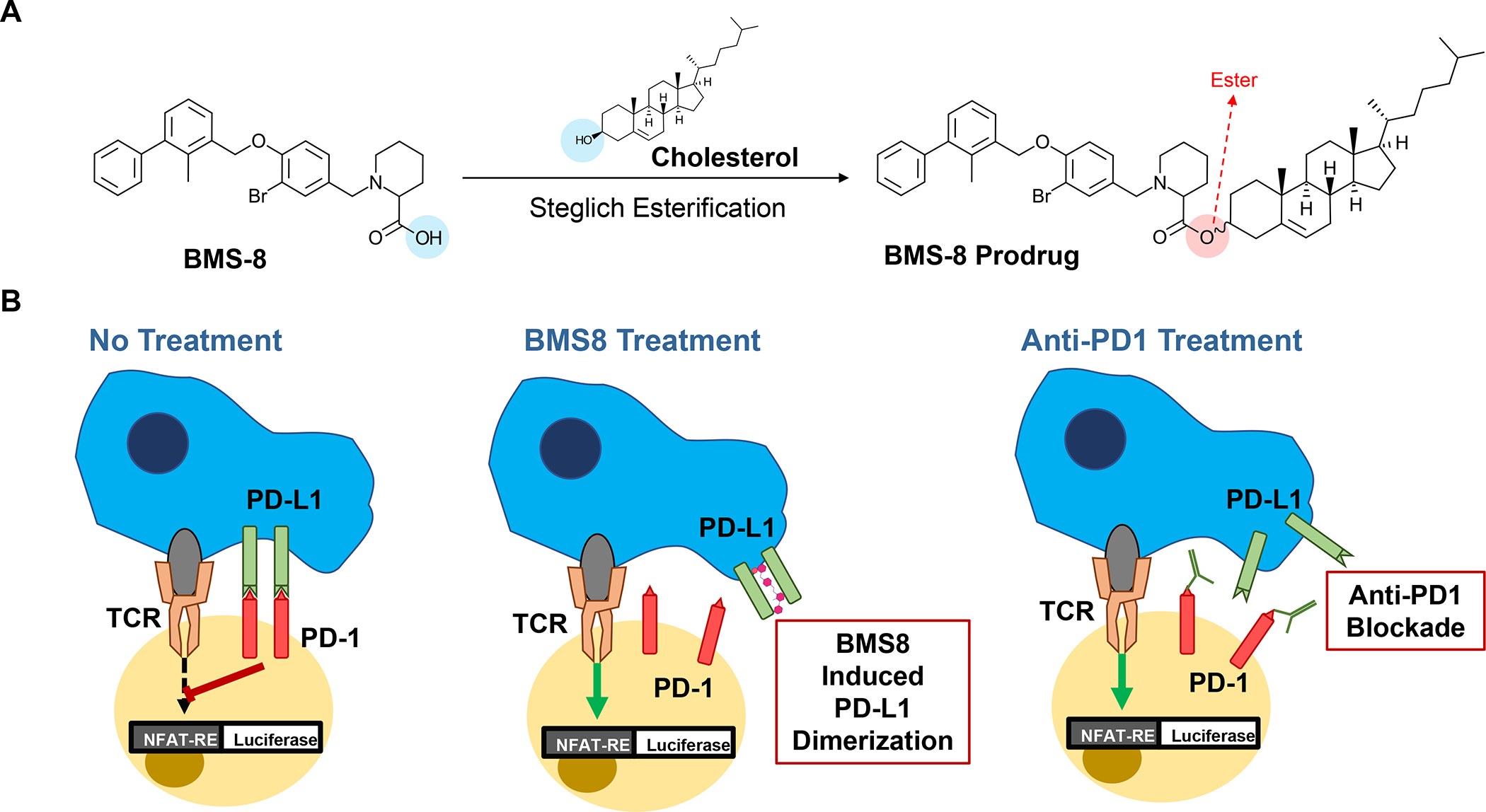
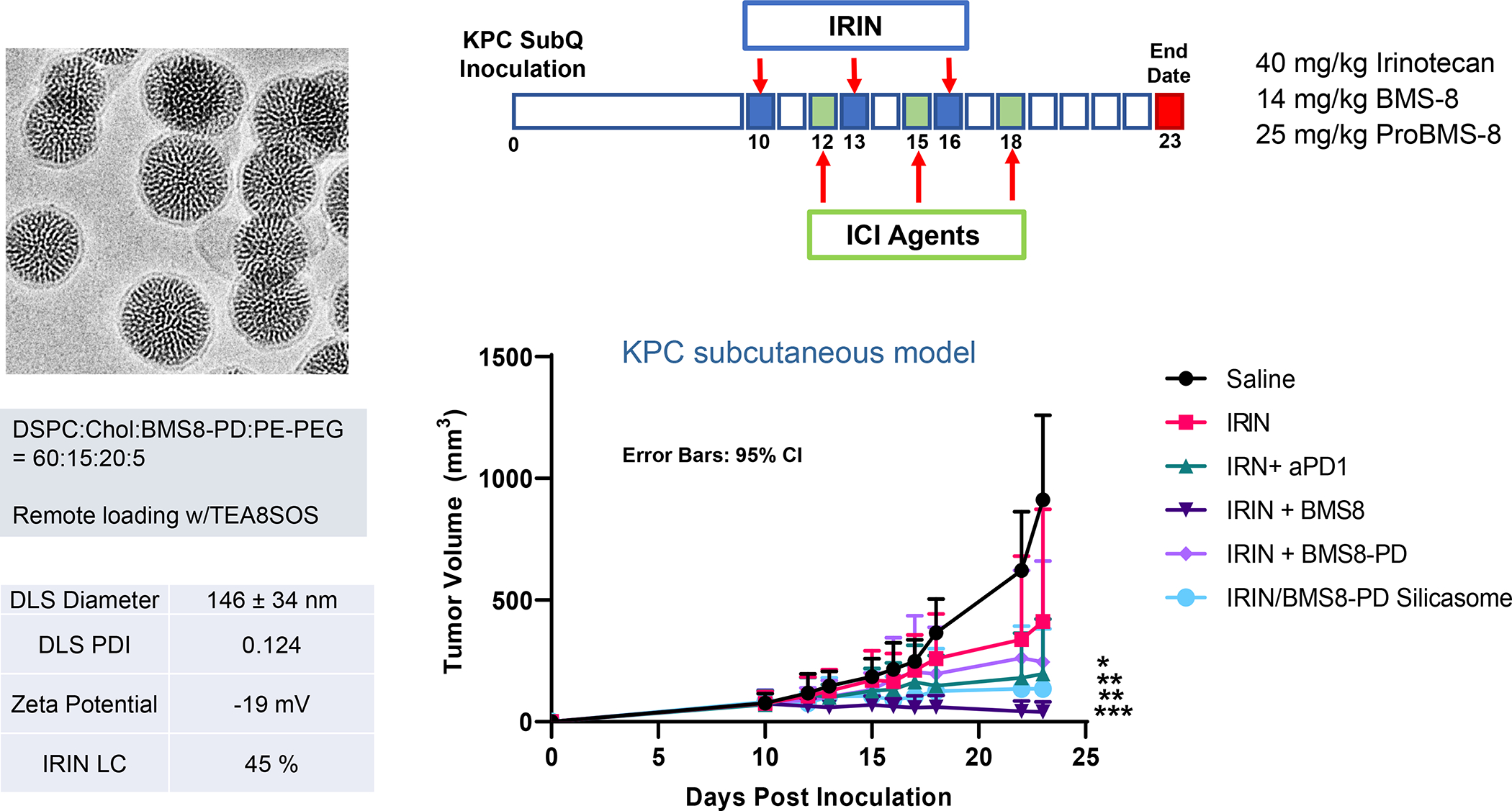
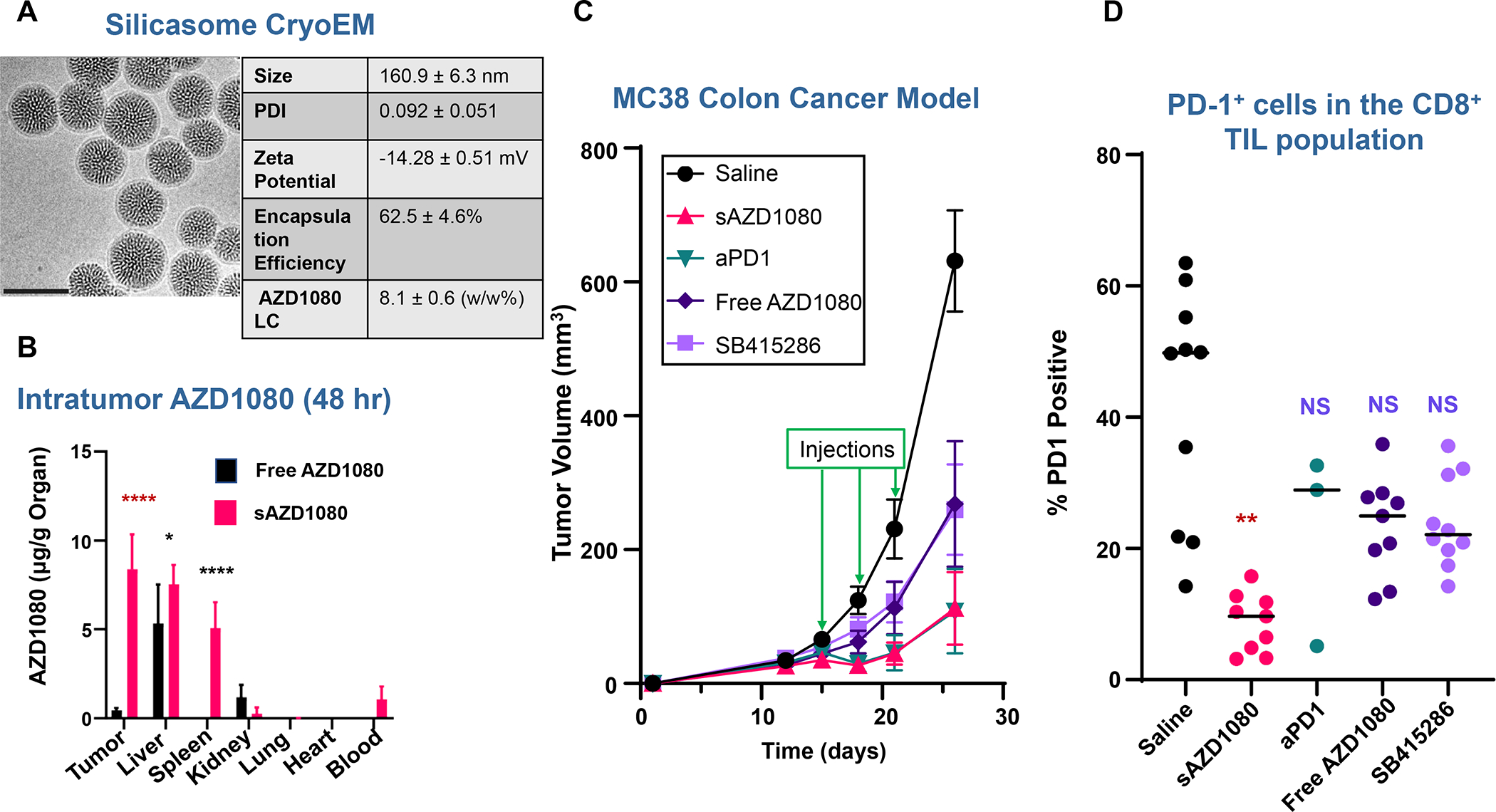
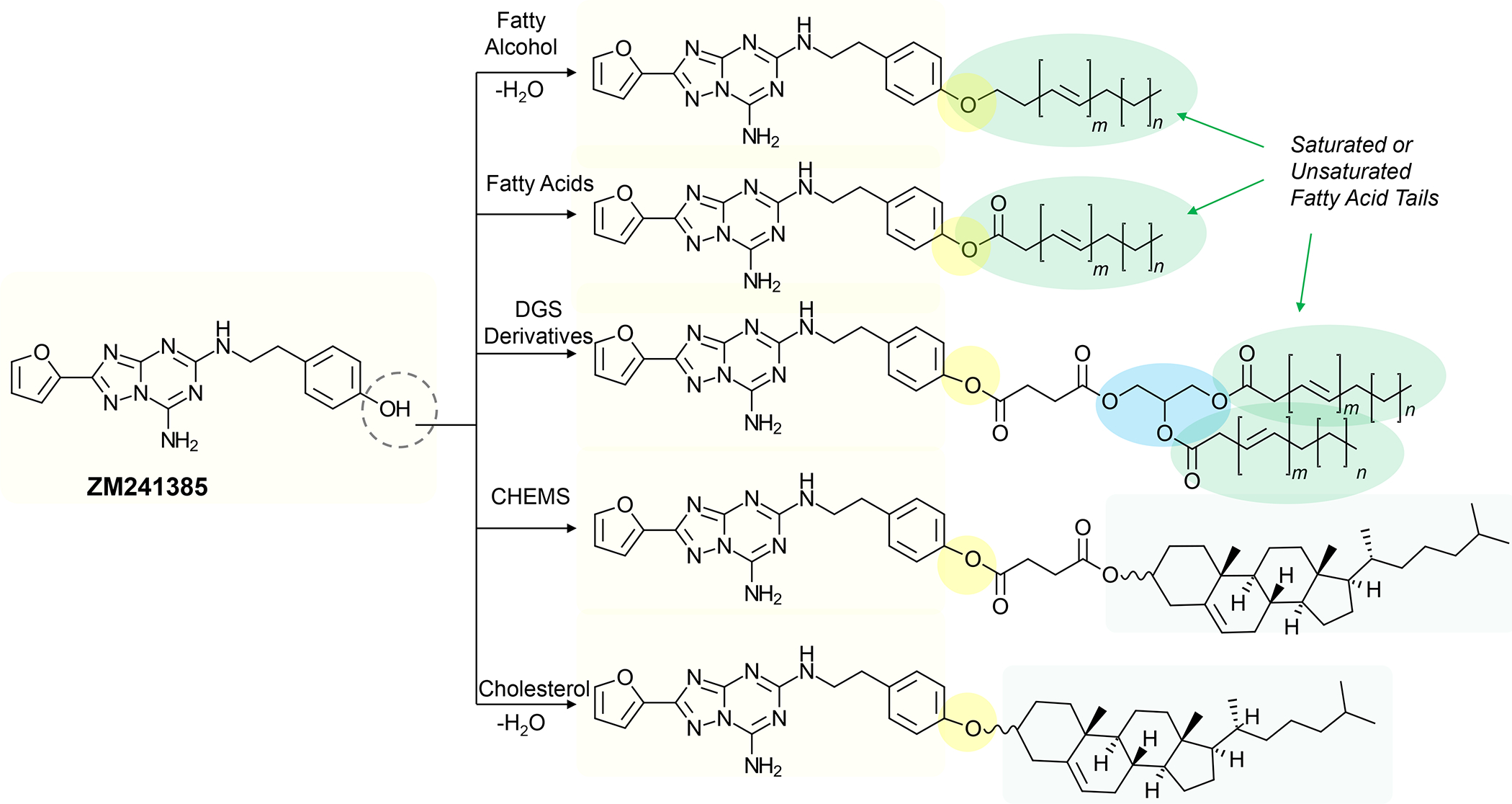
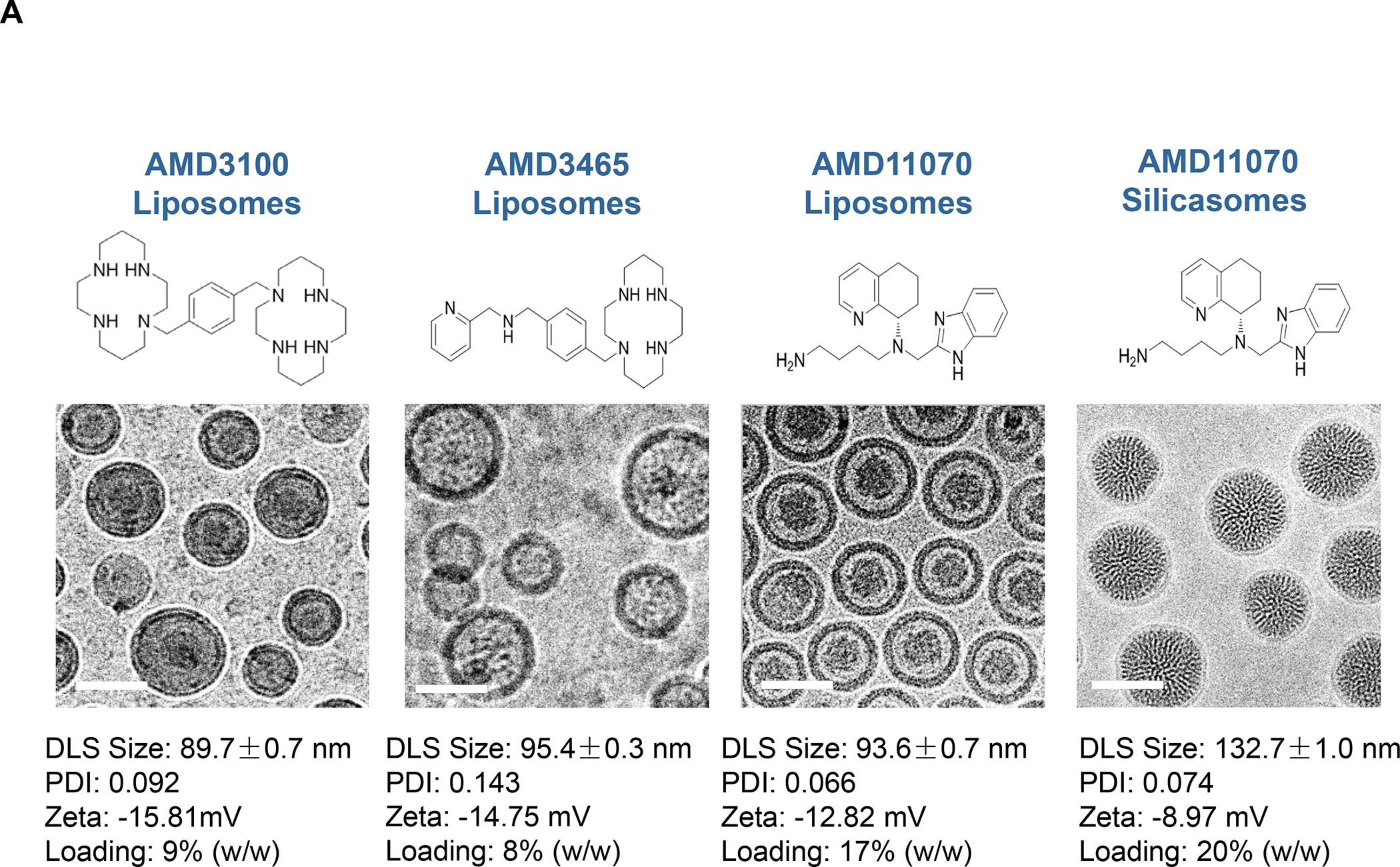
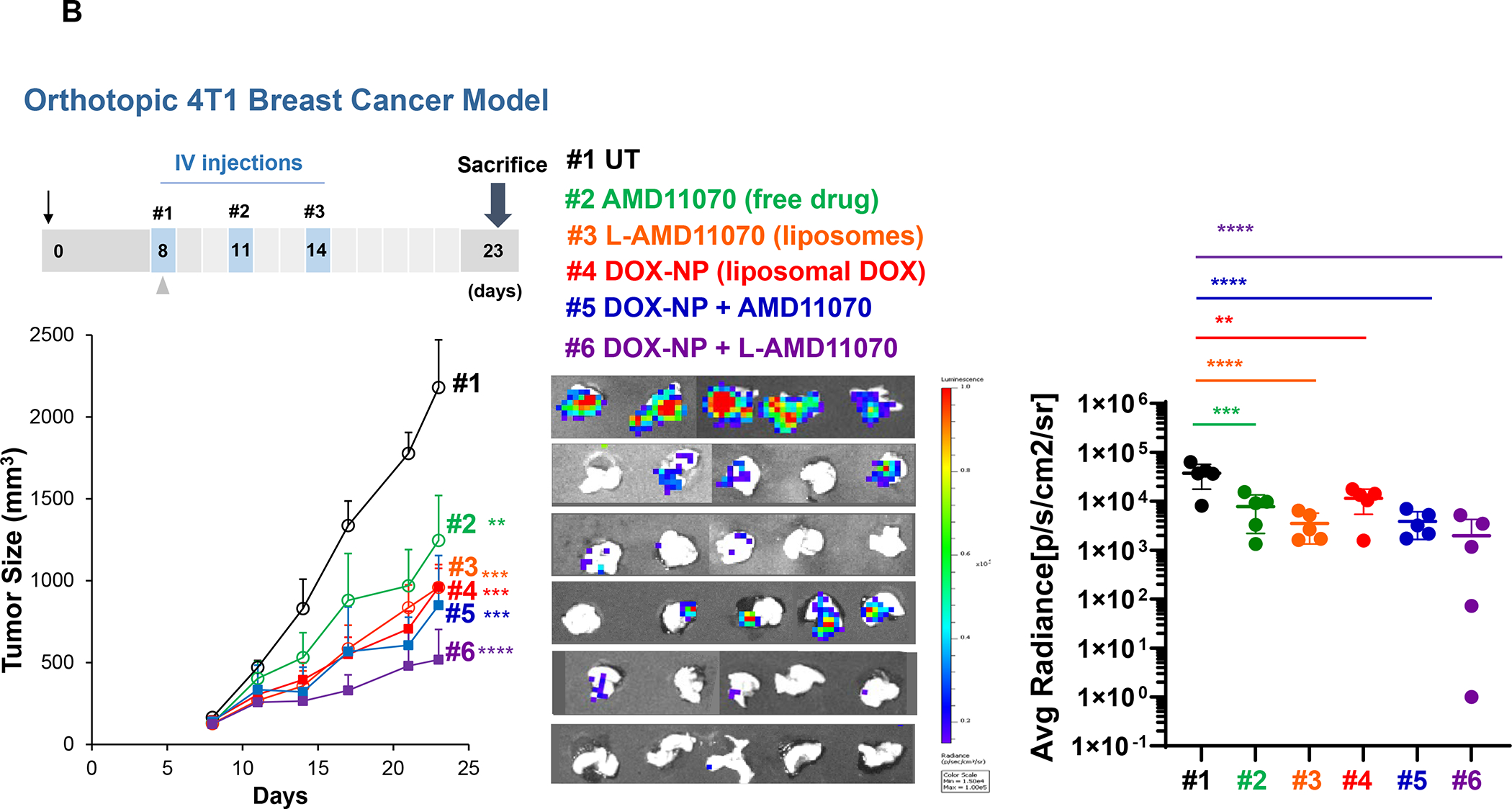
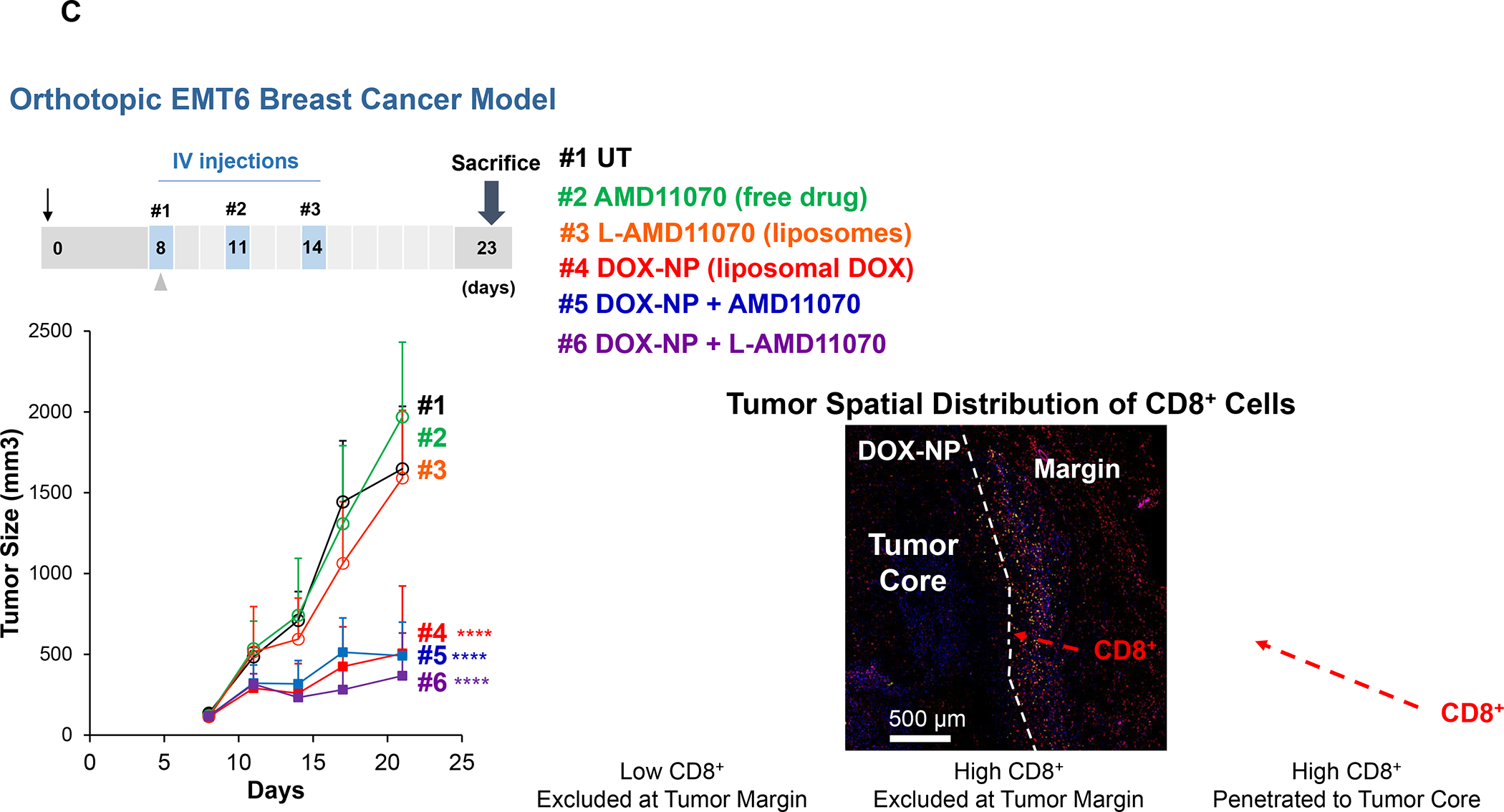
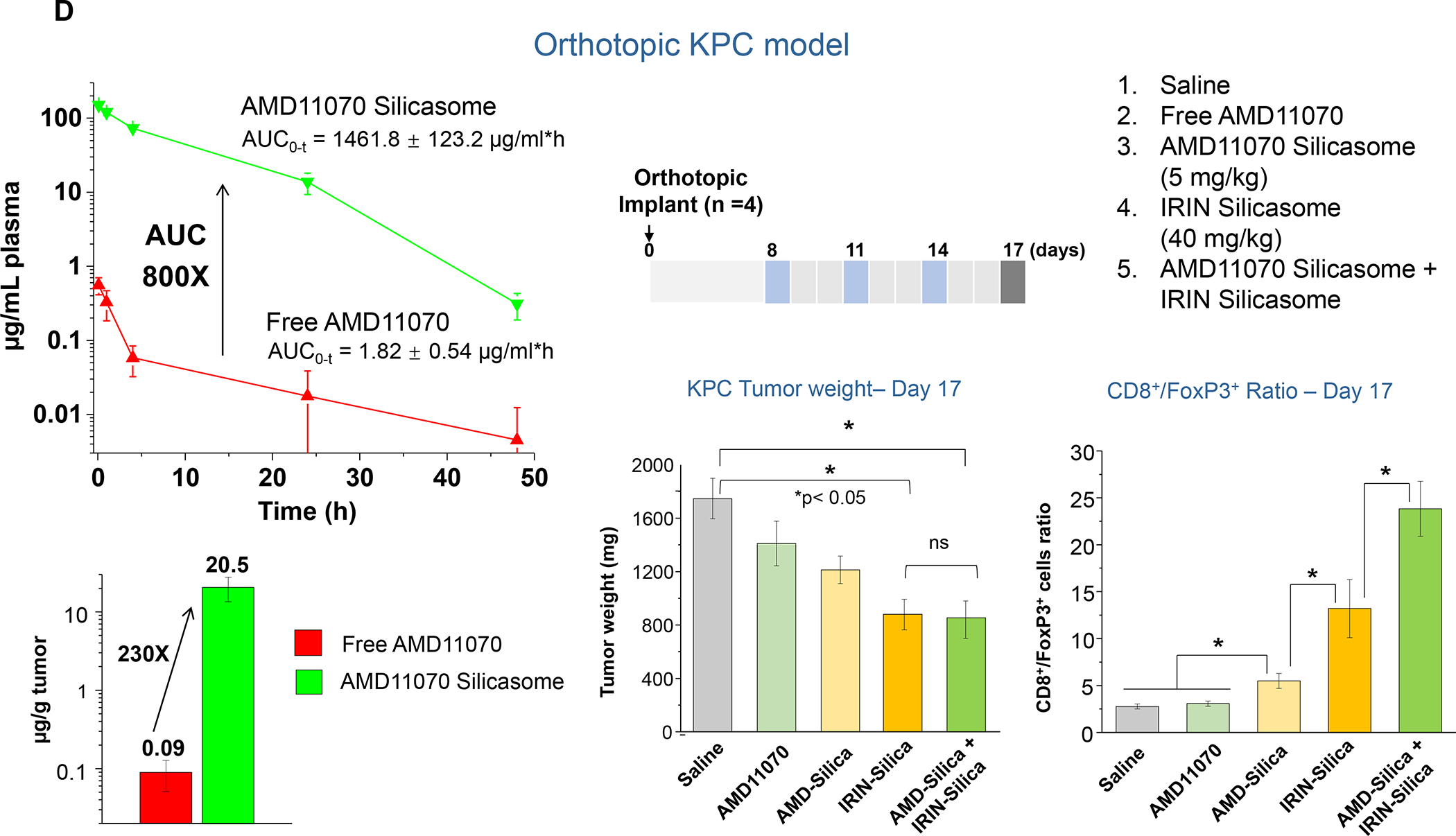
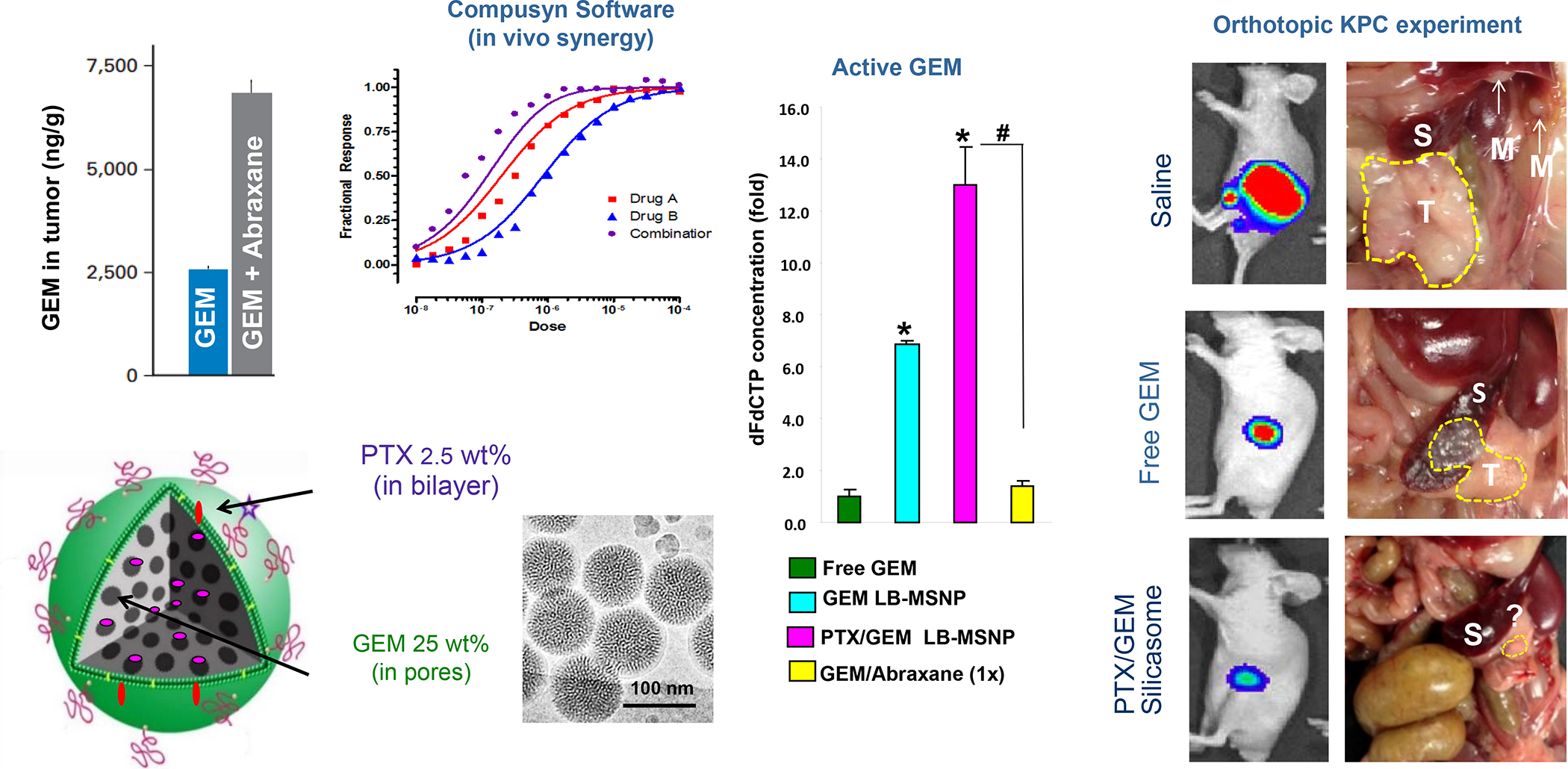
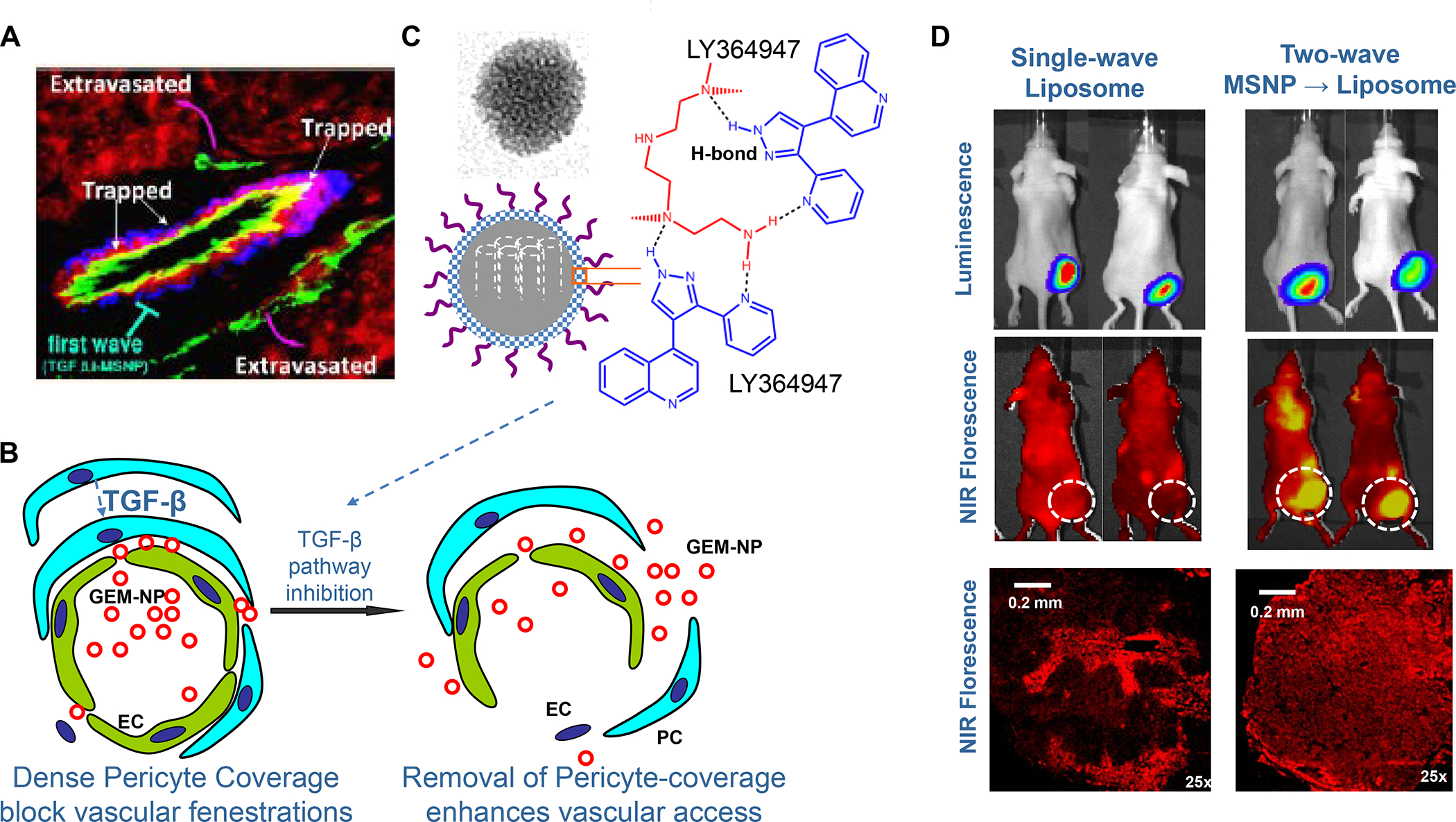
In addition to the contribution of cancer cells, the solid tumor microenvironment (TME) has a critical role in determining tumor expansion, antitumor immunity, and the response to immunotherapy. Understanding the details of the complex interplay between cancer cells and components of the TME provides an unprecedented opportunity to explore combination therapy for intervening in the immune landscape to improve immunotherapy outcome. One approach is the introduction of multifunctional nanocarriers, capable of delivering drug combinations that provide immunogenic stimuli for improvement of tumor antigen presentation, contemporaneous with the delivery of coformulated drug or synthetic molecules that provide immune danger signals or interfere in immune-escape, immune-suppressive, and T-cell exclusion pathways. This forward-looking review will discuss the use of lipid-bilayer-encapsulated liposomes and mesoporous silica nanoparticles for combination immunotherapy of the heterogeneous immune landscapes in pancreatic ductal adenocarcinoma and triple-negative breast cancer. We describe how the combination of remote drug loading and lipid bilayer encapsulation is used for the synthesis of synergistic drug combinations that induce immunogenic cell death, interfere in the PD-1/PD-L1 axis, inhibit the indoleamine-pyrrole 2,3-dioxygenase (IDO-1) immune metabolic pathway, restore spatial access to activated T-cells to the cancer site, or reduce the impact of immunosuppressive stromal components. We show how an integration of current knowledge and future discovery can be used for a rational approach to nanoenabled cancer immunotherapy.
Keywords: combination therapy; immune escape; immune landscapes; immune suppression; immunogenic cell death; liposomes; nanocarrier; pancreas cancer; silicasomes; spatial distribution; triple-negative breast cancer.
Conflict of interest statement
Competing Financial Interests
Andre E. Nel is co-founder and equity holder in Westwood Biosciences Inc. and NAMMI Therapeutics. Nel also serves on the Board for Westwood Biosciences Inc. The remaining authors declare no conflicts of interest.
Figures

Similar articles
-
Breast Cancer Chemo-immunotherapy through Liposomal Delivery of an Immunogenic Cell Death Stimulus Plus Interference in the IDO-1 Pathway.ACS Nano. 2018 Nov 27;12(11):11041-11061. doi: 10.1021/acsnano.8b05189. Epub 2018 Oct 16. ACS Nano. 2018. Retraction in: ACS Nano. 2021 Jun 22;15(6):10735. doi: 10.1021/acsnano.0c08653. PMID: 30481959 Free PMC article. Retracted.
-
Nanocarrier Co-formulation for Delivery of a TLR7 Agonist plus an Immunogenic Cell Death Stimulus Triggers Effective Pancreatic Cancer Chemo-immunotherapy.ACS Nano. 2022 Aug 23;16(8):13168-13182. doi: 10.1021/acsnano.2c06300. Epub 2022 Aug 3. ACS Nano. 2022. PMID: 35920660 Free PMC article.
-
Redox-Activated Porphyrin-Based Liposome Remote-Loaded with Indoleamine 2,3-Dioxygenase (IDO) Inhibitor for Synergistic Photoimmunotherapy through Induction of Immunogenic Cell Death and Blockage of IDO Pathway.Nano Lett. 2019 Oct 9;19(10):6964-6976. doi: 10.1021/acs.nanolett.9b02306. Epub 2019 Sep 24. Nano Lett. 2019. PMID: 31518149
-
Chemo-Immunotherapy: Role of Indoleamine 2,3-Dioxygenase in Defining Immunogenic Versus Tolerogenic Cell Death in the Tumor Microenvironment.Adv Exp Med Biol. 2017;1036:91-104. doi: 10.1007/978-3-319-67577-0_7. Adv Exp Med Biol. 2017. PMID: 29275467 Free PMC article. Review.
-
Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities.Front Immunol. 2020 Nov 5;11:598877. doi: 10.3389/fimmu.2020.598877. eCollection 2020. Front Immunol. 2020. PMID: 33250900 Free PMC article. Review.
Cited by
-
Lipid-based nanosystems: the next generation of cancer immune therapy.J Hematol Oncol. 2024 Jul 19;17(1):53. doi: 10.1186/s13045-024-01574-1. J Hematol Oncol. 2024. PMID: 39030582 Free PMC article. Review.
-
Stealth Nanocarriers in Cancer Therapy: a Comprehensive Review of Design, Functionality, and Clinical Applications.AAPS PharmSciTech. 2024 Jun 18;25(6):140. doi: 10.1208/s12249-024-02843-5. AAPS PharmSciTech. 2024. PMID: 38890191 Review.
-
Co-delivery of gemcitabine and paclitaxel plus NanoCpG empowers chemoimmunotherapy of postoperative "cold" triple-negative breast cancer.Bioact Mater. 2023 Jan 22;25:61-72. doi: 10.1016/j.bioactmat.2023.01.014. eCollection 2023 Jul. Bioact Mater. 2023. PMID: 36733927 Free PMC article.
-
Lipid-based nanoparticles for cancer immunotherapy.Med Rev (2021). 2023 Aug 17;3(3):230-269. doi: 10.1515/mr-2023-0020. eCollection 2023 Jun. Med Rev (2021). 2023. PMID: 37789955 Free PMC article. Review.
-
HSA-nanobinders crafted from bioresponsive prodrugs for combined cancer chemoimmunotherapy-an in vitro exploration.Front Chem. 2024 Mar 27;12:1378233. doi: 10.3389/fchem.2024.1378233. Front Chem. 2024. PMID: 38591056 Free PMC article.
References
-
- Yang J; Zhang C, Regulation of cancer-immunity cycle and tumor microenvironment by nanobiomaterials to enhance tumor immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1612. - PubMed
-
- Rodallec A; Sicard G; Fanciullino R; Benzekry S; Lacarelle B; Milano G; Ciccolini J, Turning Cold Tumors into Hot Tumors: Harnessing the Potential of Tumor Immunity Using Nanoparticles. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 1139–1147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
