

Propagation and Dissemination Strategies of Transmissible Spongiform Encephalopathy Agents in Mammalian Cells
- PMID: 35328330
- PMCID: PMC8949484
- DOI: 10.3390/ijms23062909
Propagation and Dissemination Strategies of Transmissible Spongiform Encephalopathy Agents in Mammalian Cells
Abstract
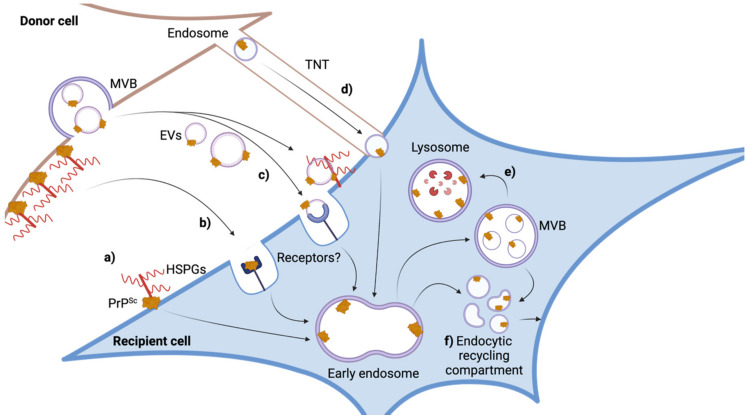
Transmissible spongiform encephalopathies or prion disorders are fatal infectious diseases that cause characteristic spongiform degeneration in the central nervous system. The causative agent, the so-called prion, is an unconventional infectious agent that propagates by converting the host-encoded cellular prion protein PrP into ordered protein aggregates with infectious properties. Prions are devoid of coding nucleic acid and thus rely on the host cell machinery for propagation. While it is now established that, in addition to PrP, other cellular factors or processes determine the susceptibility of cell lines to prion infection, exact factors and cellular processes remain broadly obscure. Still, cellular models have uncovered important aspects of prion propagation and revealed intercellular dissemination strategies shared with other intracellular pathogens. Here, we summarize what we learned about the processes of prion invasion, intracellular replication and subsequent dissemination from ex vivo cell models.
Keywords: PrP; amyloid; prion; transmissible spongiform encephalopathies; virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Cellular aspects of prion replication in vitro.Viruses. 2013 Jan 22;5(1):374-405. doi: 10.3390/v5010374. Viruses. 2013. PMID: 23340381 Free PMC article. Review.
-
The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies.FEBS J. 2007 Feb;274(3):588-605. doi: 10.1111/j.1742-4658.2007.05631.x. FEBS J. 2007. PMID: 17288548 Review.
-
Transmission and Replication of Prions.Prog Mol Biol Transl Sci. 2017;150:181-201. doi: 10.1016/bs.pmbts.2017.06.014. Epub 2017 Aug 7. Prog Mol Biol Transl Sci. 2017. PMID: 28838661 Review.
-
Prion protein self-interactions: a gateway to novel therapeutic strategies?Vaccine. 2010 Nov 16;28(49):7810-23. doi: 10.1016/j.vaccine.2010.09.012. Epub 2010 Oct 20. Vaccine. 2010. PMID: 20932496 Review.
-
[Unconventional transmissible agents or prions].Presse Med. 1997 Mar 22;26(9):425-30. Presse Med. 1997. PMID: 9137406 Review. French.
Cited by
-
Fluorimetric Detection of Insulin Misfolding by Probes Derived from Functionalized Fluorene Frameworks.Molecules. 2024 Mar 7;29(6):1196. doi: 10.3390/molecules29061196. Molecules. 2024. PMID: 38542833 Free PMC article.
-
Extracellular vesicles: A new paradigm in understanding, diagnosing and treating neurodegenerative disease.Front Aging Neurosci. 2022 Nov 3;14:967231. doi: 10.3389/fnagi.2022.967231. eCollection 2022. Front Aging Neurosci. 2022. PMID: 36408114 Free PMC article. Review.
-
Editorial: Protein Phase Separation and Aggregation in (Patho)Physiology of Neurons.Front Physiol. 2022 Jul 4;13:959570. doi: 10.3389/fphys.2022.959570. eCollection 2022. Front Physiol. 2022. PMID: 35860657 Free PMC article. No abstract available.
-
Overview of the Special Issue "Protein-Based Infection, Inheritance, and Memory".Int J Mol Sci. 2023 Jul 10;24(14):11280. doi: 10.3390/ijms241411280. Int J Mol Sci. 2023. PMID: 37511040 Free PMC article.
-
What is the role of lipids in prion conversion and disease?Front Mol Neurosci. 2023 Jan 10;15:1032541. doi: 10.3389/fnmol.2022.1032541. eCollection 2022. Front Mol Neurosci. 2023. PMID: 36704327 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
