

The role of PI3Kγ in the immune system: new insights and translational implications
- PMID: 35322259
- PMCID: PMC9922156
- DOI: 10.1038/s41577-022-00701-8
The role of PI3Kγ in the immune system: new insights and translational implications
Abstract
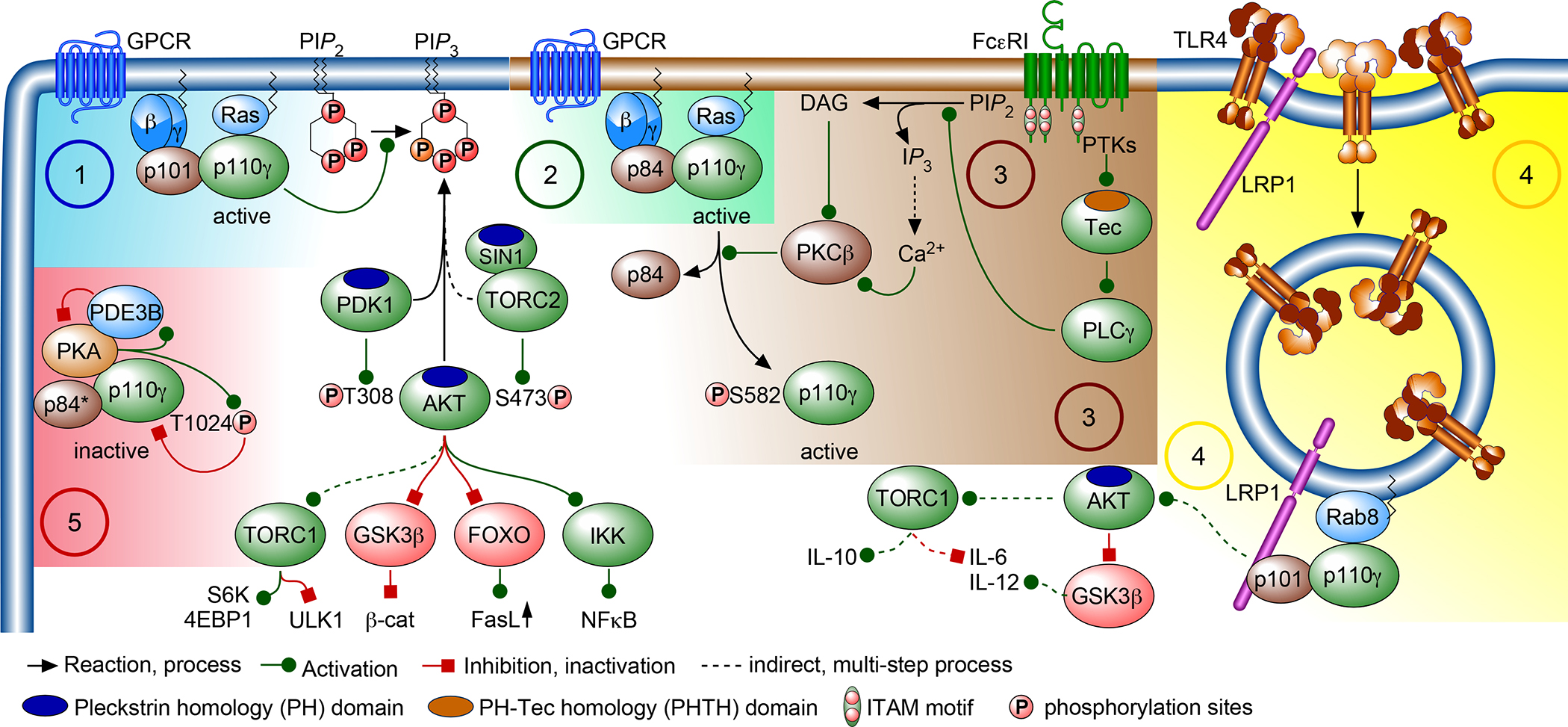
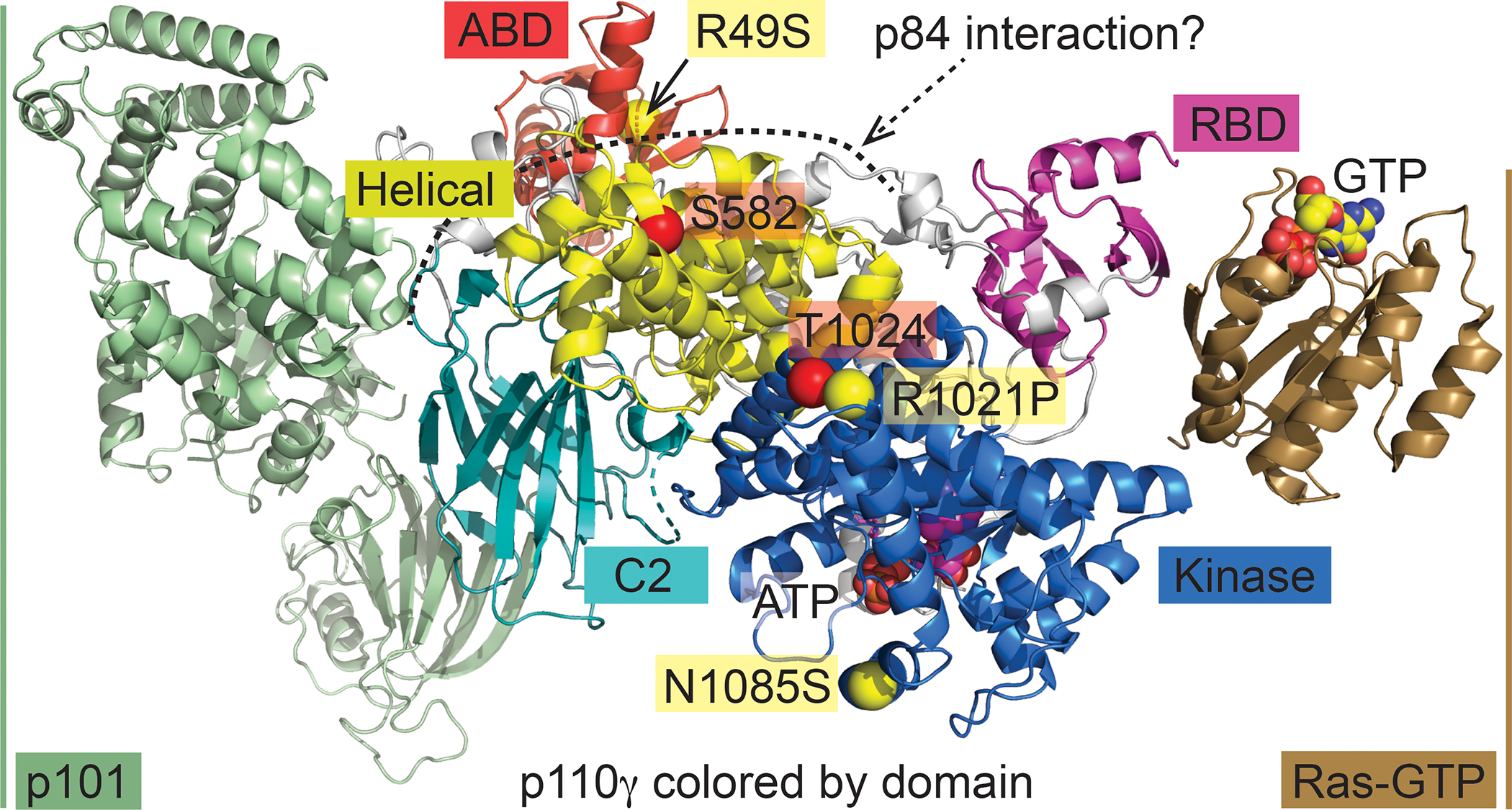
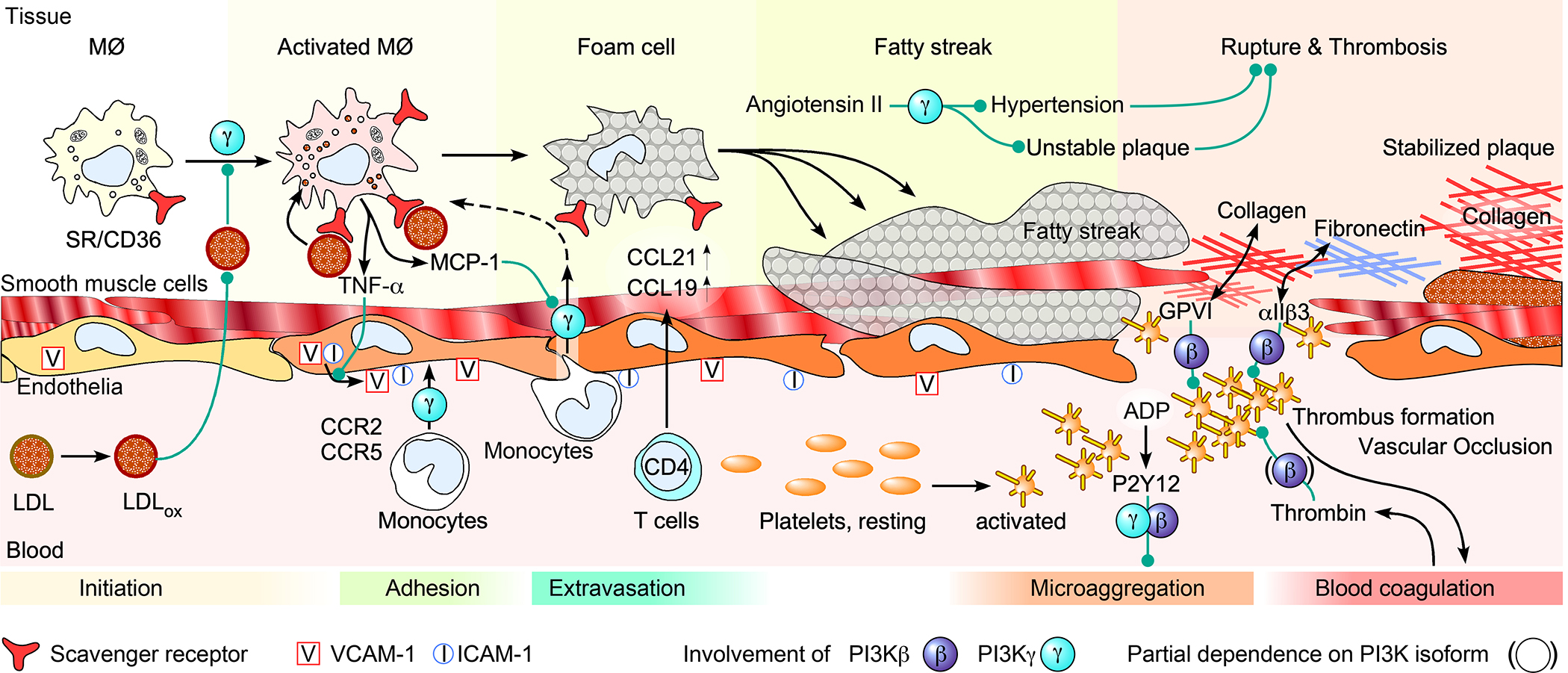
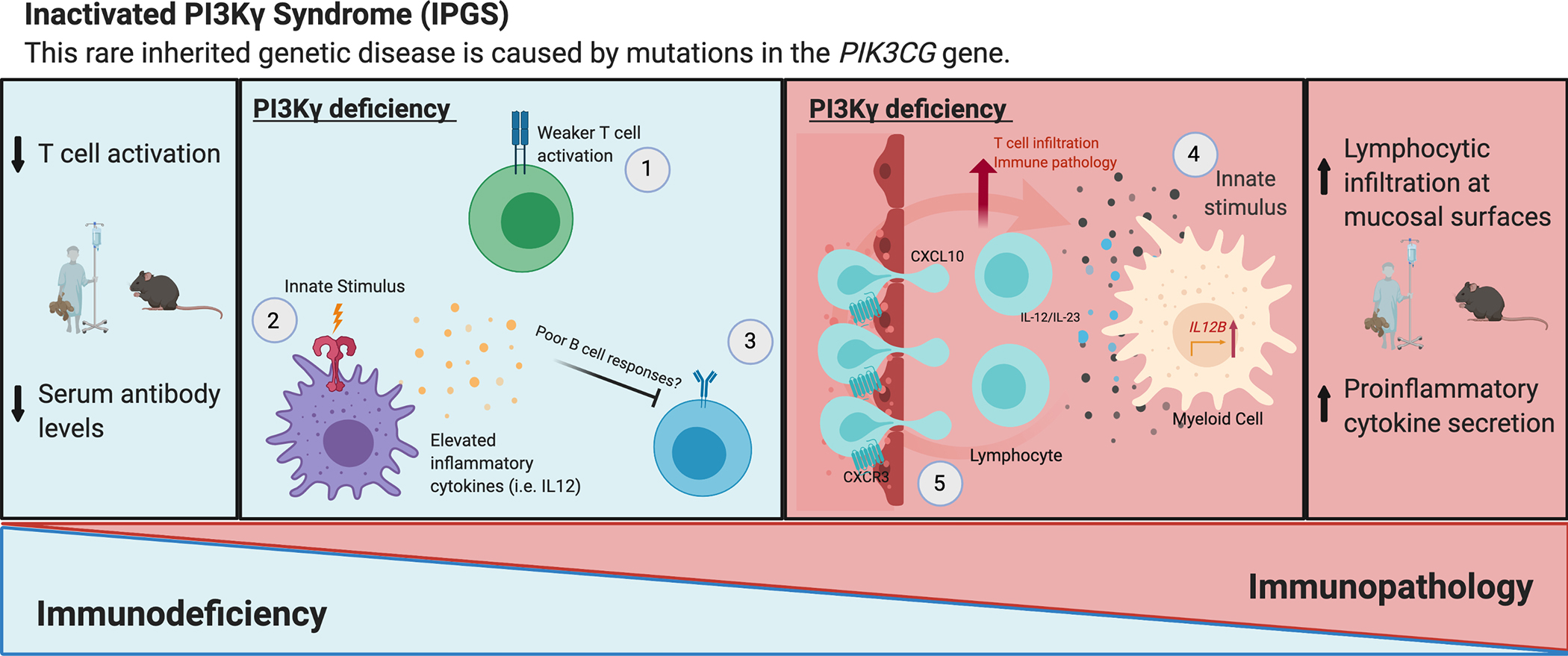
Over the past two decades, new insights have positioned phosphoinositide 3-kinase-γ (PI3Kγ) as a context-dependent modulator of immunity and inflammation. Recent advances in protein structure determination and drug development have allowed for generation of highly specific PI3Kγ inhibitors, with the first now in clinical trials for several oncology indications. Recently, a monogenic immune disorder caused by PI3Kγ deficiency was discovered in humans and modelled in mice. Human inactivated PI3Kγ syndrome confirms the immunomodulatory roles of PI3Kγ and strengthens newly defined roles of this molecule in modulating inflammatory cytokine release in macrophages. Here, we review the functions of PI3Kγ in the immune system and discuss how our understanding of its potential as a therapeutic target has evolved.
© 2022. Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Similar articles
-
Targeting phosphatidylinositol 3-kinase gamma (PI3Kγ): Discovery and development of its selective inhibitors.Med Res Rev. 2021 May;41(3):1599-1621. doi: 10.1002/med.21770. Epub 2020 Dec 10. Med Res Rev. 2021. PMID: 33300614 Review.
-
Phosphoinositide 3 Kinase γ Plays a Critical Role in Acute Kidney Injury.Cells. 2022 Feb 23;11(5):772. doi: 10.3390/cells11050772. Cells. 2022. PMID: 35269396 Free PMC article.
-
Designing Small Molecule PI3Kγ Inhibitors: A Review of Structure-Based Methods and Computational Approaches.J Med Chem. 2024 Jul 11;67(13):10530-10547. doi: 10.1021/acs.jmedchem.4c00347. Epub 2024 Jun 19. J Med Chem. 2024. PMID: 38988222 Review.
-
A class of highly selective inhibitors bind to an active state of PI3Kγ.Nat Chem Biol. 2019 Apr;15(4):348-357. doi: 10.1038/s41589-018-0215-0. Epub 2019 Feb 4. Nat Chem Biol. 2019. PMID: 30718815
-
Evolution of PI3Kγ and δ Inhibitors for Inflammatory and Autoimmune Diseases.J Med Chem. 2019 May 23;62(10):4783-4814. doi: 10.1021/acs.jmedchem.8b01298. Epub 2018 Dec 24. J Med Chem. 2019. PMID: 30582813 Review.
Cited by
-
Eganelisib combined with immune checkpoint inhibitor therapy and chemotherapy in frontline metastatic triple-negative breast cancer triggers macrophage reprogramming, immune activation and extracellular matrix reorganization in the tumor microenvironment.J Immunother Cancer. 2024 Aug 30;12(8):e009160. doi: 10.1136/jitc-2024-009160. J Immunother Cancer. 2024. PMID: 39214650 Free PMC article. Clinical Trial.
-
Molecular basis for differential activation of p101 and p84 complexes of PI3Kγ by Ras and GPCRs.Cell Rep. 2023 Mar 28;42(3):112172. doi: 10.1016/j.celrep.2023.112172. Epub 2023 Feb 26. Cell Rep. 2023. PMID: 36842083 Free PMC article.
-
Antioxidant and Wound Healing Bioactive Potential of Extracts Obtained from Bark and Needles of Softwood Species.Antioxidants (Basel). 2023 Jul 4;12(7):1383. doi: 10.3390/antiox12071383. Antioxidants (Basel). 2023. PMID: 37507922 Free PMC article.
-
Allosteric activation or inhibition of PI3Kγ mediated through conformational changes in the p110γ helical domain.bioRxiv [Preprint]. 2023 May 23:2023.04.12.536585. doi: 10.1101/2023.04.12.536585. bioRxiv. 2023. Update in: Elife. 2023 Jul 07;12:RP88058. doi: 10.7554/eLife.88058 PMID: 37090531 Free PMC article. Updated. Preprint.
-
The immune system as a driver of mitochondrial disease pathogenesis: a review of evidence.Orphanet J Rare Dis. 2022 Sep 2;17(1):335. doi: 10.1186/s13023-022-02495-3. Orphanet J Rare Dis. 2022. PMID: 36056365 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
