

Essential role of hyperacetylated microtubules in innate immunity escape orchestrated by the EBV-encoded BHRF1 protein
- PMID: 35275978
- PMCID: PMC8942261
- DOI: 10.1371/journal.ppat.1010371
Essential role of hyperacetylated microtubules in innate immunity escape orchestrated by the EBV-encoded BHRF1 protein
Abstract
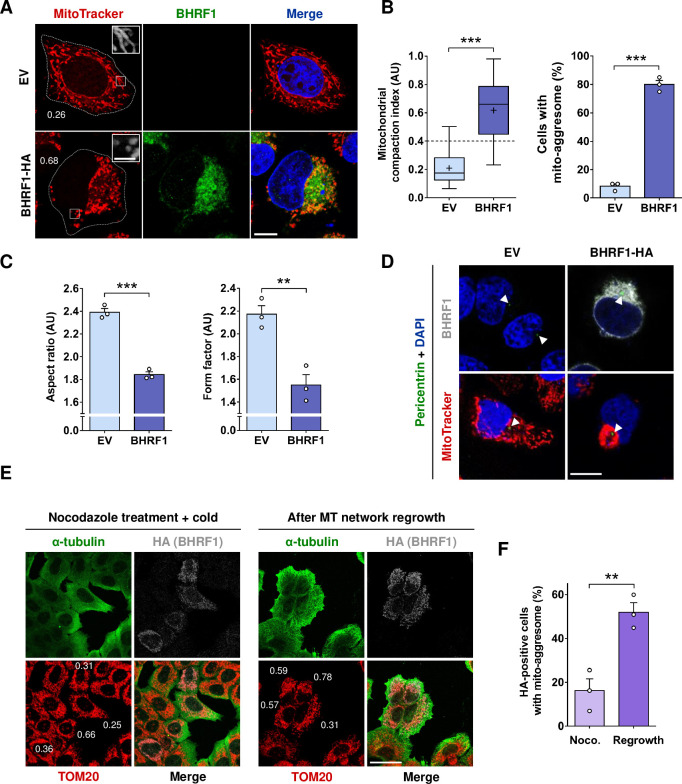
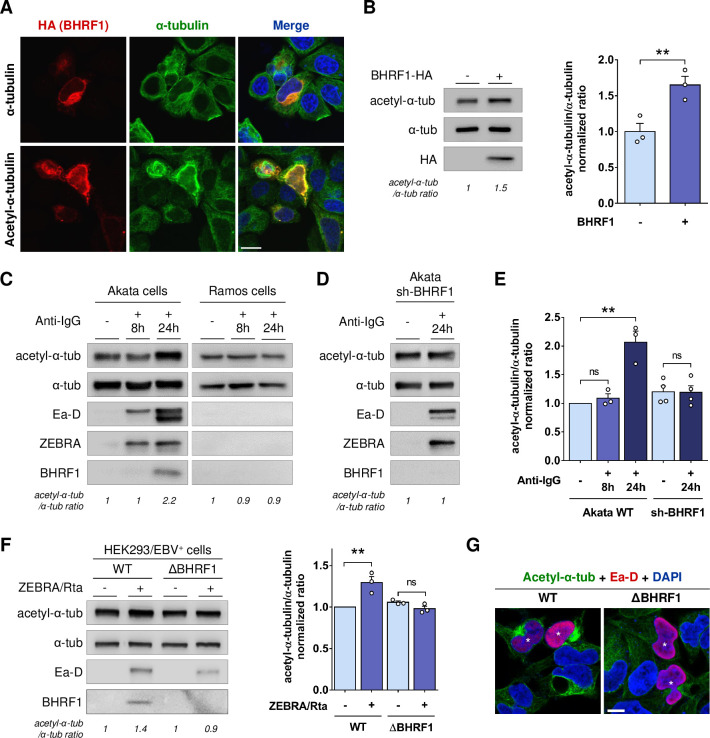
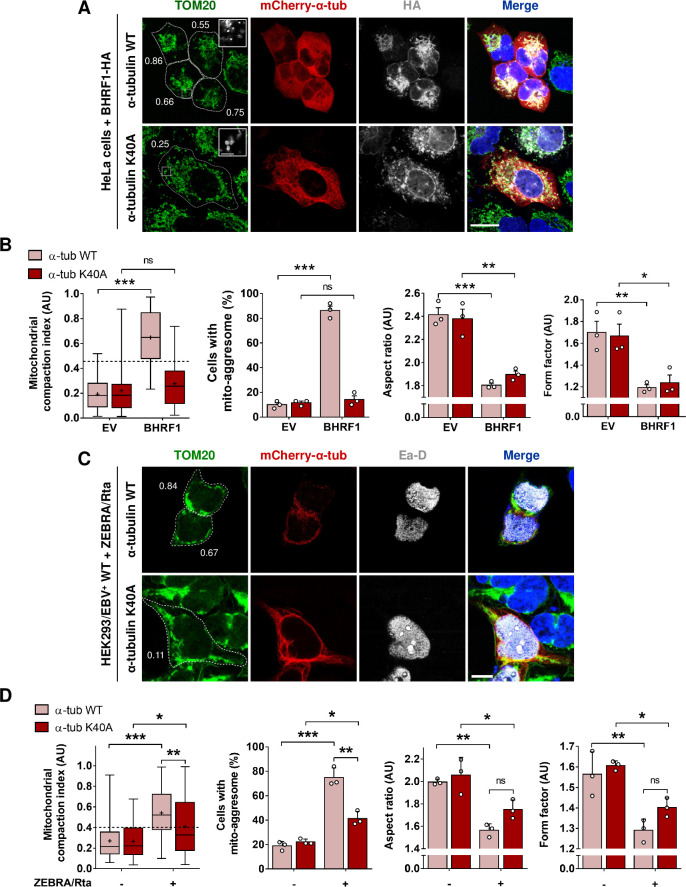
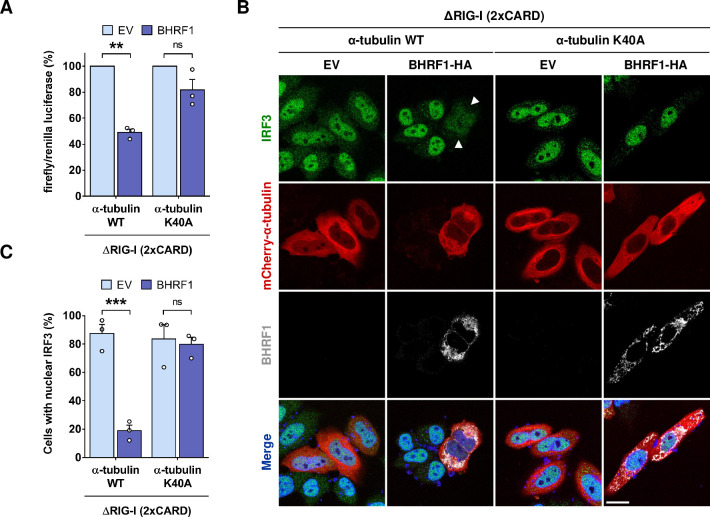
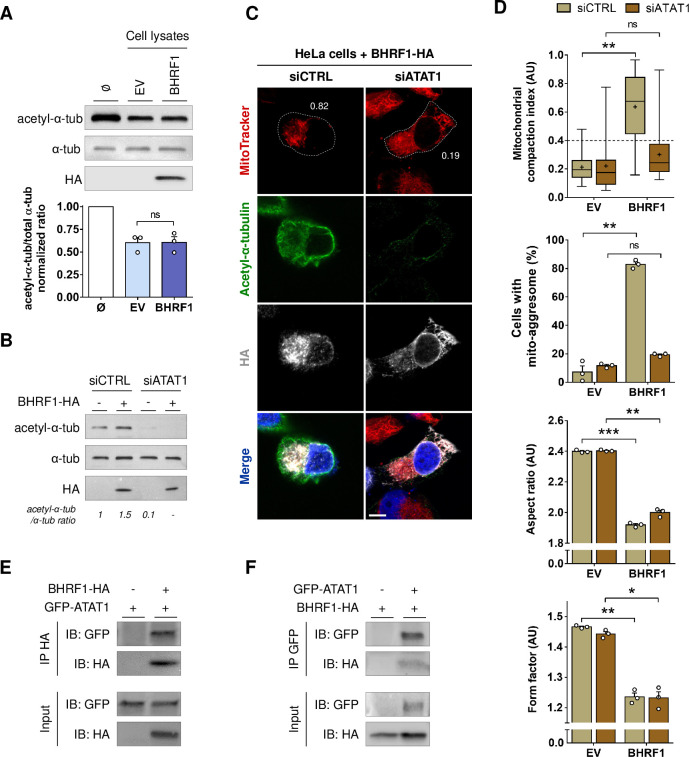
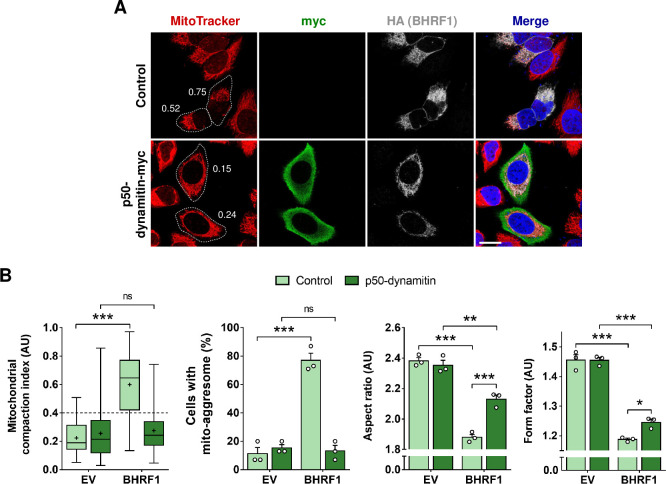
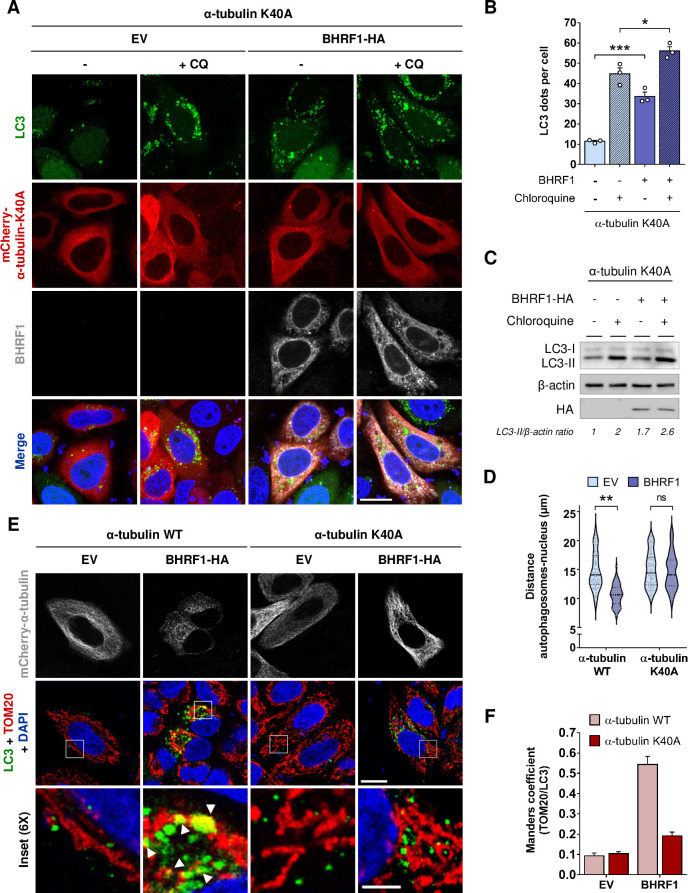
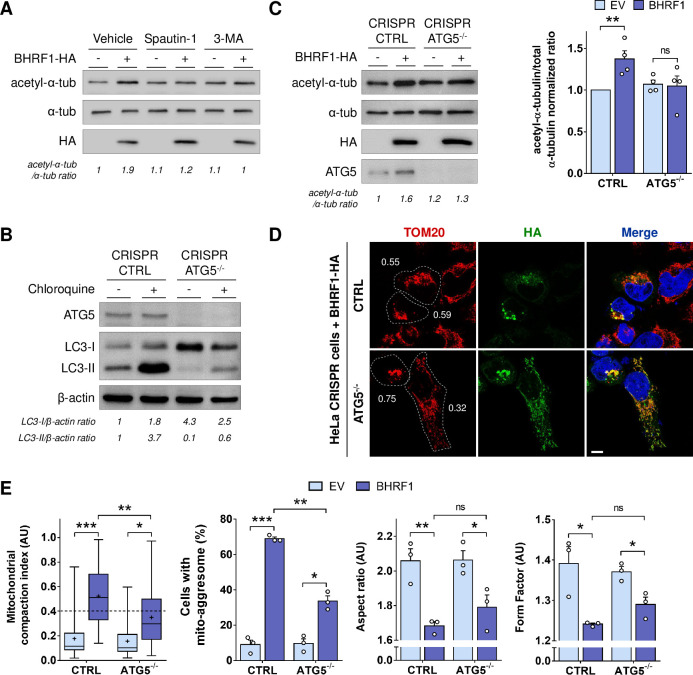
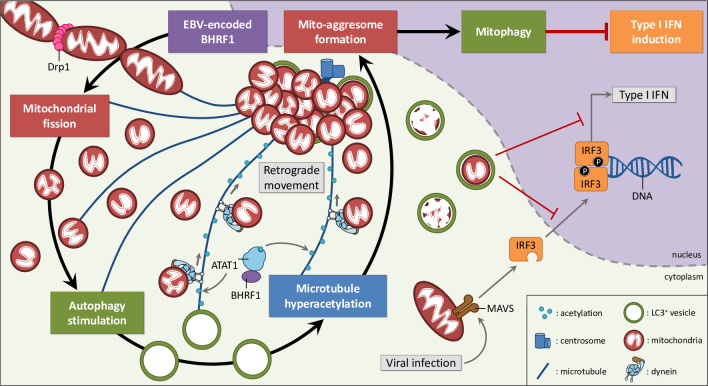
Innate immunity constitutes the first line of defense against viruses, in which mitochondria play an important role in the induction of the interferon (IFN) response. BHRF1, a multifunctional viral protein expressed during Epstein-Barr virus reactivation, modulates mitochondrial dynamics and disrupts the IFN signaling pathway. Mitochondria are mobile organelles that move through the cytoplasm thanks to the cytoskeleton and in particular the microtubule (MT) network. MTs undergo various post-translational modifications, among them tubulin acetylation. In this study, we demonstrated that BHRF1 induces MT hyperacetylation to escape innate immunity. Indeed, the expression of BHRF1 induces the clustering of shortened mitochondria next to the nucleus. This "mito-aggresome" is organized around the centrosome and its formation is MT-dependent. We also observed that the α-tubulin acetyltransferase ATAT1 interacts with BHRF1. Using ATAT1 knockdown or a non-acetylatable α-tubulin mutant, we demonstrated that this hyperacetylation is necessary for the mito-aggresome formation. Similar results were observed during EBV reactivation. We investigated the mechanism leading to the clustering of mitochondria, and we identified dyneins as motors that are required for mitochondrial clustering. Finally, we demonstrated that BHRF1 needs MT hyperacetylation to block the induction of the IFN response. Moreover, the loss of MT hyperacetylation blocks the localization of autophagosomes close to the mito-aggresome, impeding BHRF1 to initiate mitophagy, which is essential to inhibiting the signaling pathway. Therefore, our results reveal the role of the MT network, and its acetylation level, in the induction of a pro-viral mitophagy.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction.Autophagy. 2021 Jun;17(6):1296-1315. doi: 10.1080/15548627.2020.1758416. Epub 2020 May 13. Autophagy. 2021. PMID: 32401605 Free PMC article.
-
BHRF1 Enhances EBV Mediated Nasopharyngeal Carcinoma Tumorigenesis through Modulating Mitophagy Associated with Mitochondrial Membrane Permeabilization Transition.Cells. 2020 May 7;9(5):1158. doi: 10.3390/cells9051158. Cells. 2020. PMID: 32392902 Free PMC article.
-
An Epstein-Barr Virus MicroRNA Blocks Interleukin-1 (IL-1) Signaling by Targeting IL-1 Receptor 1.J Virol. 2017 Oct 13;91(21):e00530-17. doi: 10.1128/JVI.00530-17. Print 2017 Nov 1. J Virol. 2017. PMID: 28794034 Free PMC article.
-
Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases.Front Immunol. 2020 Mar 3;11:367. doi: 10.3389/fimmu.2020.00367. eCollection 2020. Front Immunol. 2020. PMID: 32194570 Free PMC article. Review.
-
Direct and indirect effects of tubulin post-translational modifications on microtubule stability: Insights and regulations.Biochim Biophys Acta Mol Cell Res. 2022 Jun;1869(6):119241. doi: 10.1016/j.bbamcr.2022.119241. Epub 2022 Feb 15. Biochim Biophys Acta Mol Cell Res. 2022. PMID: 35181405 Review.
Cited by
-
Vimentin inhibits α-tubulin acetylation via enhancing α-TAT1 degradation to suppress the replication of human parainfluenza virus type 3.PLoS Pathog. 2022 Sep 15;18(9):e1010856. doi: 10.1371/journal.ppat.1010856. eCollection 2022 Sep. PLoS Pathog. 2022. PMID: 36108090 Free PMC article.
-
Tubulin Post-Translational Modifications: The Elusive Roles of Acetylation.Biology (Basel). 2023 Apr 6;12(4):561. doi: 10.3390/biology12040561. Biology (Basel). 2023. PMID: 37106761 Free PMC article. Review.
-
Crosstalk between Autophagy and RLR Signaling.Cells. 2023 Mar 21;12(6):956. doi: 10.3390/cells12060956. Cells. 2023. PMID: 36980296 Free PMC article. Review.
-
Hyperacetylated microtubules assist porcine deltacoronavirus nsp8 to degrade MDA5 via SQSTM1/p62-dependent selective autophagy.J Virol. 2024 Mar 19;98(3):e0000324. doi: 10.1128/jvi.00003-24. Epub 2024 Feb 14. J Virol. 2024. PMID: 38353538 Free PMC article.
-
Functional Implications of Epstein-Barr Virus Lytic Genes in Carcinogenesis.Cancers (Basel). 2022 Nov 24;14(23):5780. doi: 10.3390/cancers14235780. Cancers (Basel). 2022. PMID: 36497262 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
