

T1AM Attenuates the Hypoxia/Reoxygenation-Induced Necroptosis of H9C2 Cardiomyocytes via RIPK1/RIPK3 Pathway
- PMID: 35265713
- PMCID: PMC8901330
- DOI: 10.1155/2022/4833791
T1AM Attenuates the Hypoxia/Reoxygenation-Induced Necroptosis of H9C2 Cardiomyocytes via RIPK1/RIPK3 Pathway
Abstract
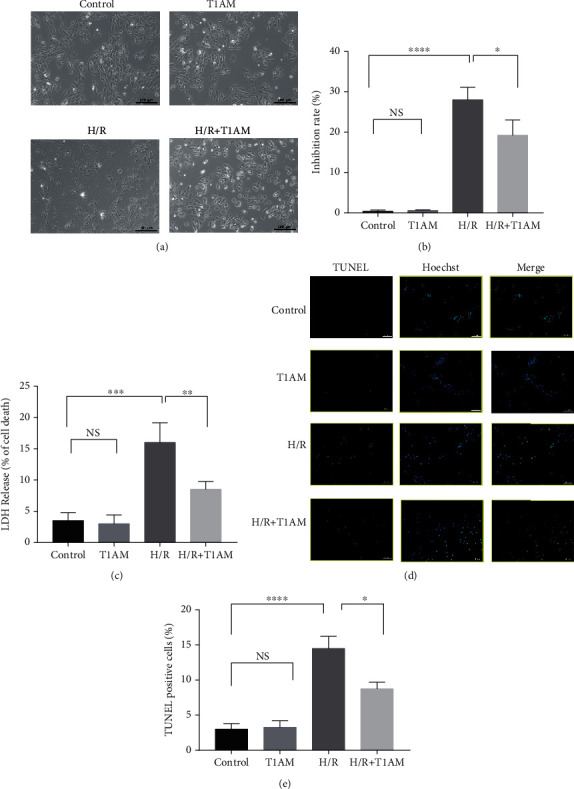
Purpose: To investigate the detailed mechanism of 3-iodothyronamine (T1AM) in cell apoptosis and programmed necrosis of hypoxia/reoxygenation- (H/R-) induced H9C2 injury.
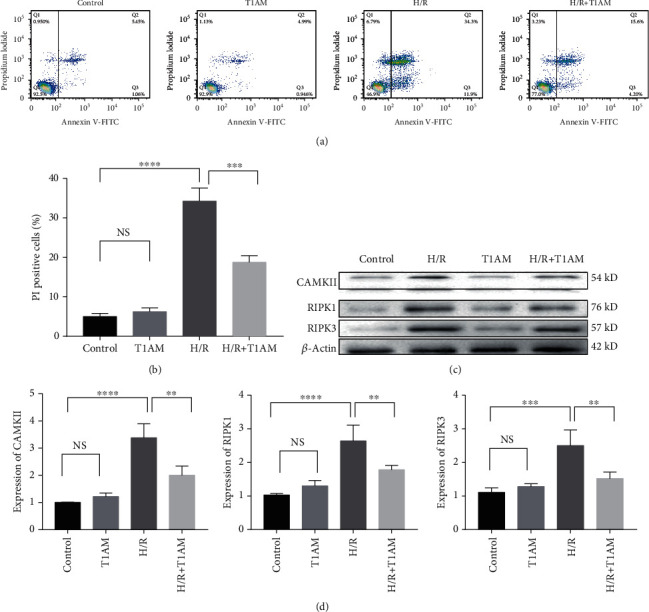
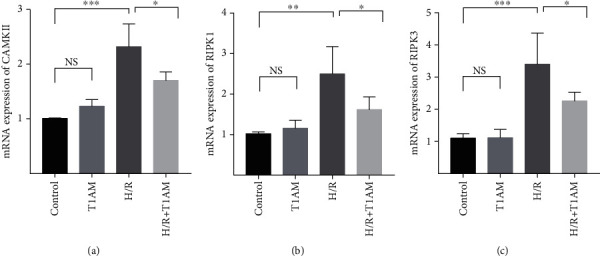
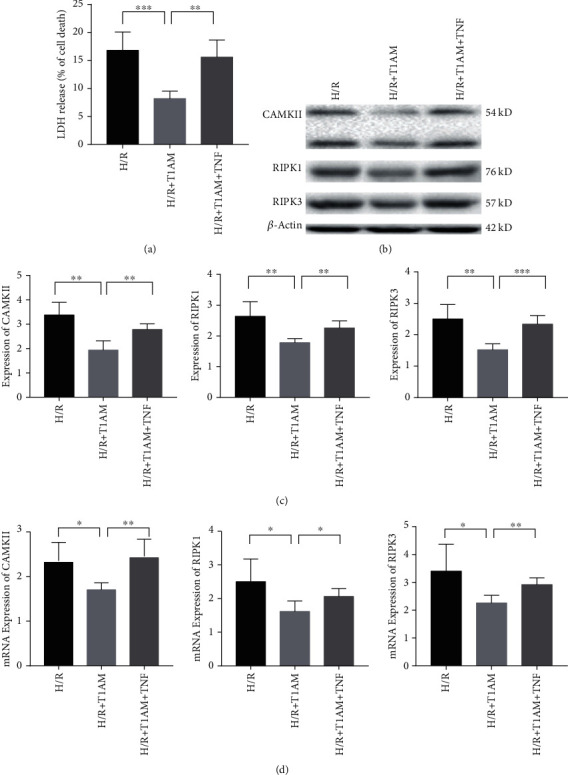
Materials and methods: Cardiomyocyte H9C2 cells were cultured in vitro for the establishment of cardiomyocyte H/R models. Cells were randomly divided into four groups: the control group, H/R group, T1AM pretreatment group, T1AM pretreatment and H/R (6 μm T1AM+H/R) group. The degree of myocardial injury was determined by the detection of the cardiomyocyte inhibition rate by CCK8 and the detection of lactic dehydrogenase (LDH) activity. Cell apoptosis was assessed through TUNEL assay and flow cytometry analysis. The protein level and mRNA level of RIPK1, RIPK3, and CAMKII were detected by western blotting and qRT-PCR.
Results: Compared with the control group, the cell inhibition rate was dramatically elevated in the H/R group. LDH release of cardiomyocytes was significantly increased. Protein and mRNA expressions of RIPK1, RIPK3, and CAMKII were significantly enhanced. Compared with the H/R group, the cell inhibition rate, LDH release, cardiomyocyte necroptosis rate, and protein and mRNA levels of RIPK1, RIPK3, and CAMKII of the T1AM+H/R group were significantly decreased.
Conclusion: Pretreatment with T1AM could alleviate cardiomyocytes' H/R injury and inhibit necroptosis of cardiomyocytes, which might exert a protective function upon activation of the RIPK1/RIPK3 pathway.
Copyright © 2022 Bo Wei et al.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Ca2+/Calmodulin-Dependent Protein Kinase II Regulation by Inhibitor of Receptor Interacting Protein Kinase 3 Alleviates Necroptosis in Glycation End Products-Induced Cardiomyocytes Injury.Int J Mol Sci. 2022 Jun 23;23(13):6988. doi: 10.3390/ijms23136988. Int J Mol Sci. 2022. PMID: 35805993 Free PMC article.
-
[Effects of Inhibiting Necroptosis on H9c2 Cardiomyocytes Injury Induced by Hypoxia/Reoxygenation].Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2015 Apr;32(2):393-9. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2015. PMID: 26211260 Chinese.
-
MicroRNA-325-3p protects the heart after myocardial infarction by inhibiting RIPK3 and programmed necrosis in mice.BMC Mol Biol. 2019 Jun 27;20(1):17. doi: 10.1186/s12867-019-0133-z. BMC Mol Biol. 2019. PMID: 31248365 Free PMC article.
-
The role of RIPK3-regulated cell death pathways and necroptosis in the pathogenesis of cardiac ischaemia-reperfusion injury.Acta Physiol (Oxf). 2021 Feb;231(2):e13541. doi: 10.1111/apha.13541. Epub 2020 Aug 7. Acta Physiol (Oxf). 2021. PMID: 32687661 Review.
-
RIPK1 and RIPK3: critical regulators of inflammation and cell death.Trends Cell Biol. 2015 Jun;25(6):347-53. doi: 10.1016/j.tcb.2015.01.001. Epub 2015 Feb 4. Trends Cell Biol. 2015. PMID: 25662614 Review.
Cited by
-
Role of RIPK3‑CaMKII‑mPTP signaling pathway‑mediated necroptosis in cardiovascular diseases (Review).Int J Mol Med. 2023 Oct;52(4):98. doi: 10.3892/ijmm.2023.5301. Epub 2023 Sep 1. Int J Mol Med. 2023. PMID: 37654208 Free PMC article. Review.
-
Hydrogen Sulfide Suppresses Skin Fibroblast Proliferation via Oxidative Stress Alleviation and Necroptosis Inhibition.Oxid Med Cell Longev. 2022 Jun 21;2022:7434733. doi: 10.1155/2022/7434733. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 35774378 Free PMC article.
References
-
- Paradies G., Paradies V., Ruggiero F. M., Petrosillo G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. American Journal of Physiology. Heart and Circulatory Physiology . 2018;315(5):H1341–h1352. doi: 10.1152/ajpheart.00028.2018. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
