

Conformational Variation in Enzyme Catalysis: A Structural Study on Catalytic Residues
- PMID: 35240125
- PMCID: PMC9005782
- DOI: 10.1016/j.jmb.2022.167517
Conformational Variation in Enzyme Catalysis: A Structural Study on Catalytic Residues
Abstract
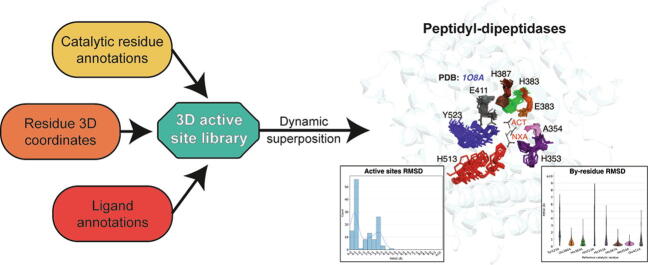
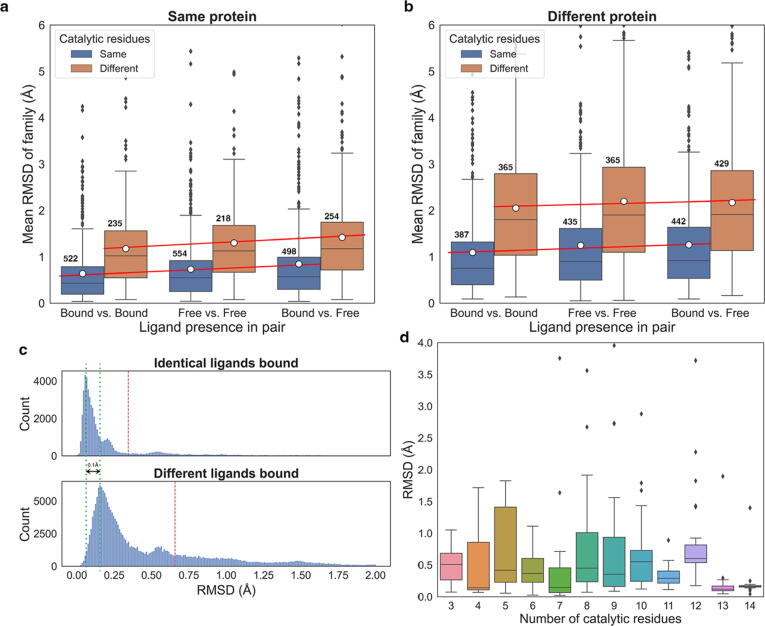
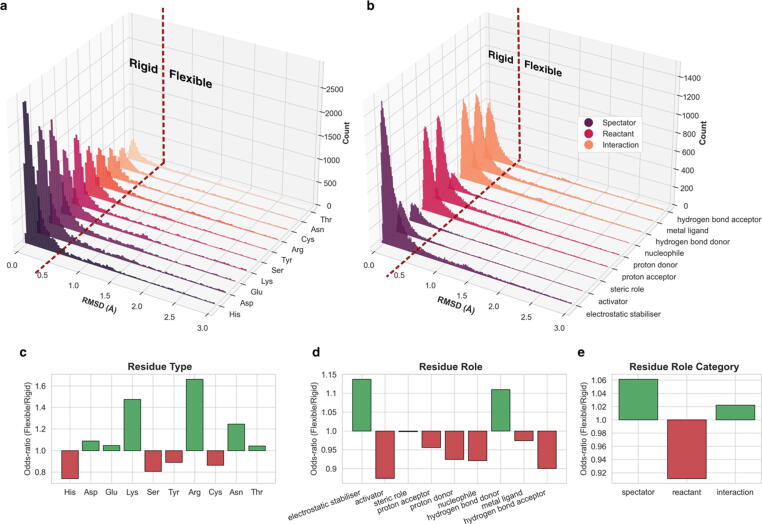
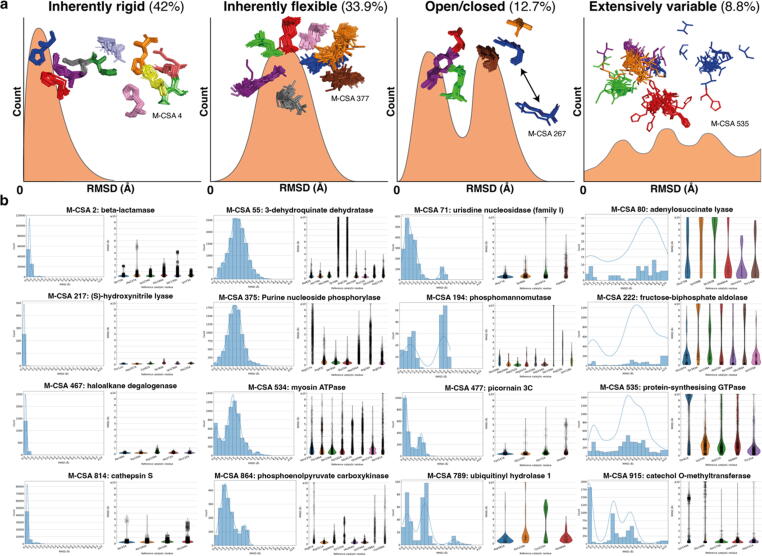
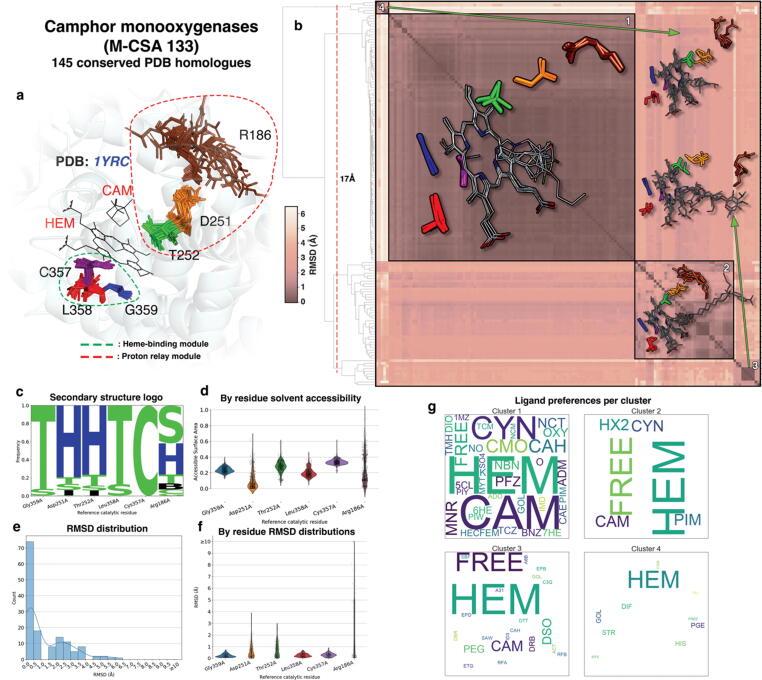
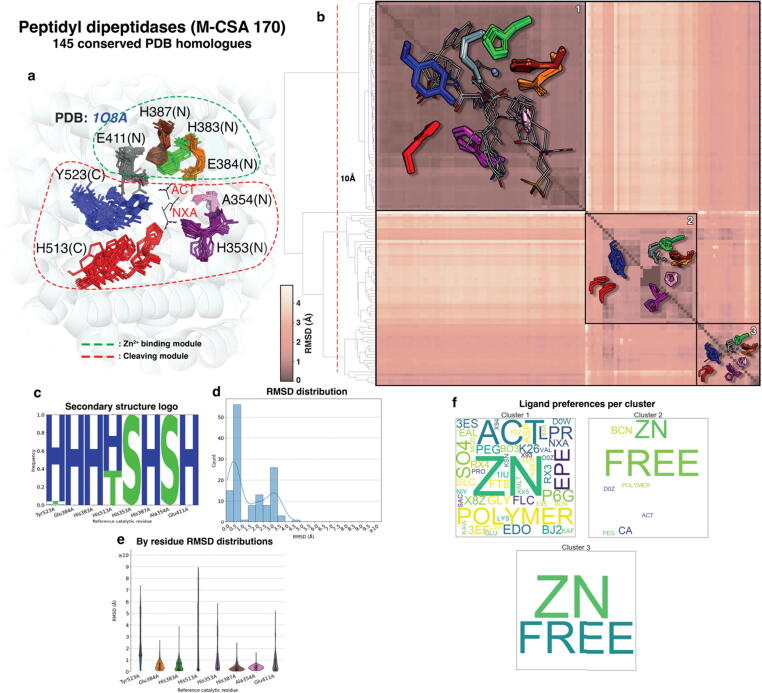
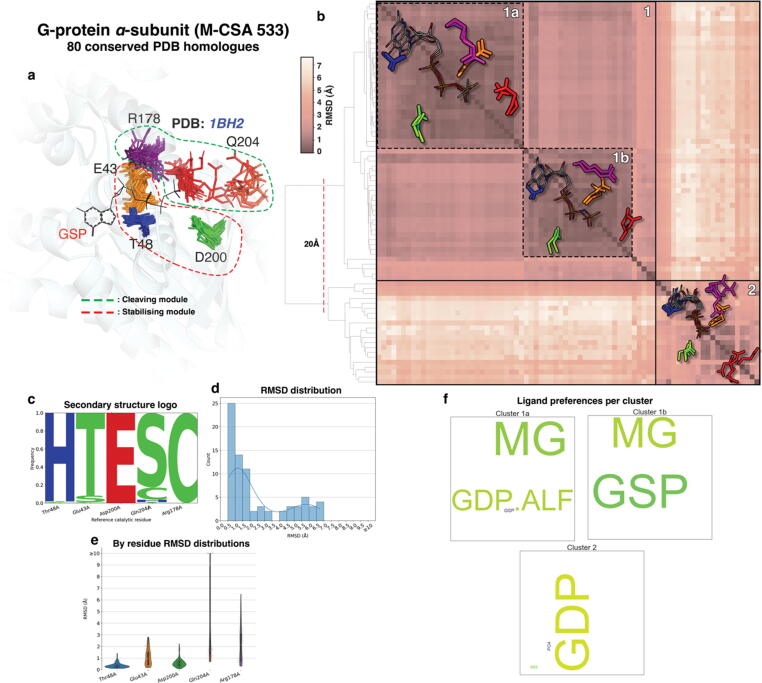
Conformational variation in catalytic residues can be captured as alternative snapshots in enzyme crystal structures. Addressing the question of whether active site flexibility is an intrinsic and essential property of enzymes for catalysis, we present a comprehensive study on the 3D variation of active sites of 925 enzyme families, using explicit catalytic residue annotations from the Mechanism and Catalytic Site Atlas and structural data from the Protein Data Bank. Through weighted pairwise superposition of the functional atoms of active sites, we captured structural variability at single-residue level and examined the geometrical changes as ligands bind or as mutations occur. We demonstrate that catalytic centres of enzymes can be inherently rigid or flexible to various degrees according to the function they perform, and structural variability most often involves a subset of the catalytic residues, usually those not directly involved in the formation or cleavage of bonds. Moreover, data suggest that 2/3 of active sites are flexible, and in half of those, flexibility is only observed in the side chain. The goal of this work is to characterise our current knowledge of the extent of flexibility at the heart of catalysis and ultimately place our findings in the context of the evolution of catalysis as enzymes evolve new functions and bind different substrates.
Keywords: Catalysis; Catalytic residues; Enzyme; Flexibility; Structure.
Copyright © 2022 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures

Similar articles
-
The Catalytic Site Atlas 2.0: cataloging catalytic sites and residues identified in enzymes.Nucleic Acids Res. 2014 Jan;42(Database issue):D485-9. doi: 10.1093/nar/gkt1243. Epub 2013 Dec 6. Nucleic Acids Res. 2014. PMID: 24319146 Free PMC article.
-
A global analysis of function and conservation of catalytic residues in enzymes.J Biol Chem. 2020 Jan 10;295(2):314-324. doi: 10.1074/jbc.REV119.006289. Epub 2019 Dec 3. J Biol Chem. 2020. PMID: 31796628 Free PMC article. Review.
-
Exploring functionally related enzymes using radially distributed properties of active sites around the reacting points of bound ligands.BMC Struct Biol. 2012 Apr 26;12:5. doi: 10.1186/1472-6807-12-5. BMC Struct Biol. 2012. PMID: 22536854 Free PMC article.
-
Looking at enzymes from the inside out: the proximity of catalytic residues to the molecular centroid can be used for detection of active sites and enzyme-ligand interfaces.J Mol Biol. 2005 Aug 12;351(2):309-26. doi: 10.1016/j.jmb.2005.06.047. J Mol Biol. 2005. PMID: 16019028
-
Active site flexibility in enzyme catalysis.Ann N Y Acad Sci. 1998 Dec 13;864:1-8. doi: 10.1111/j.1749-6632.1998.tb10282.x. Ann N Y Acad Sci. 1998. PMID: 9928078 Review.
Cited by
-
Enzyme function and evolution through the lens of bioinformatics.Biochem J. 2023 Nov 29;480(22):1845-1863. doi: 10.1042/BCJ20220405. Biochem J. 2023. PMID: 37991346 Free PMC article. Review.
-
Capturing the geometry, function, and evolution of enzymes with 3D templates.Protein Sci. 2022 Jul;31(7):e4363. doi: 10.1002/pro.4363. Protein Sci. 2022. PMID: 35762726 Free PMC article. Review.
-
A role for conformational changes in enzyme catalysis.Biophys J. 2024 Jun 18;123(12):1563-1578. doi: 10.1016/j.bpj.2024.04.030. Epub 2024 May 3. Biophys J. 2024. PMID: 38704639
-
A review on application of molecular simulation technology in food molecules interaction.Curr Res Food Sci. 2022 Oct 11;5:1873-1881. doi: 10.1016/j.crfs.2022.10.012. eCollection 2022. Curr Res Food Sci. 2022. PMID: 36276243 Free PMC article. Review.
-
Catalytic site flexibility facilitates the substrate and catalytic promiscuity of Vibrio dual lipase/transferase.Nat Commun. 2023 Aug 9;14(1):4795. doi: 10.1038/s41467-023-40455-y. Nat Commun. 2023. PMID: 37558668 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
