

Spatial organization of transcribed eukaryotic genes
- PMID: 35177821
- PMCID: PMC9380065
- DOI: 10.1038/s41556-022-00847-6
Spatial organization of transcribed eukaryotic genes
Abstract
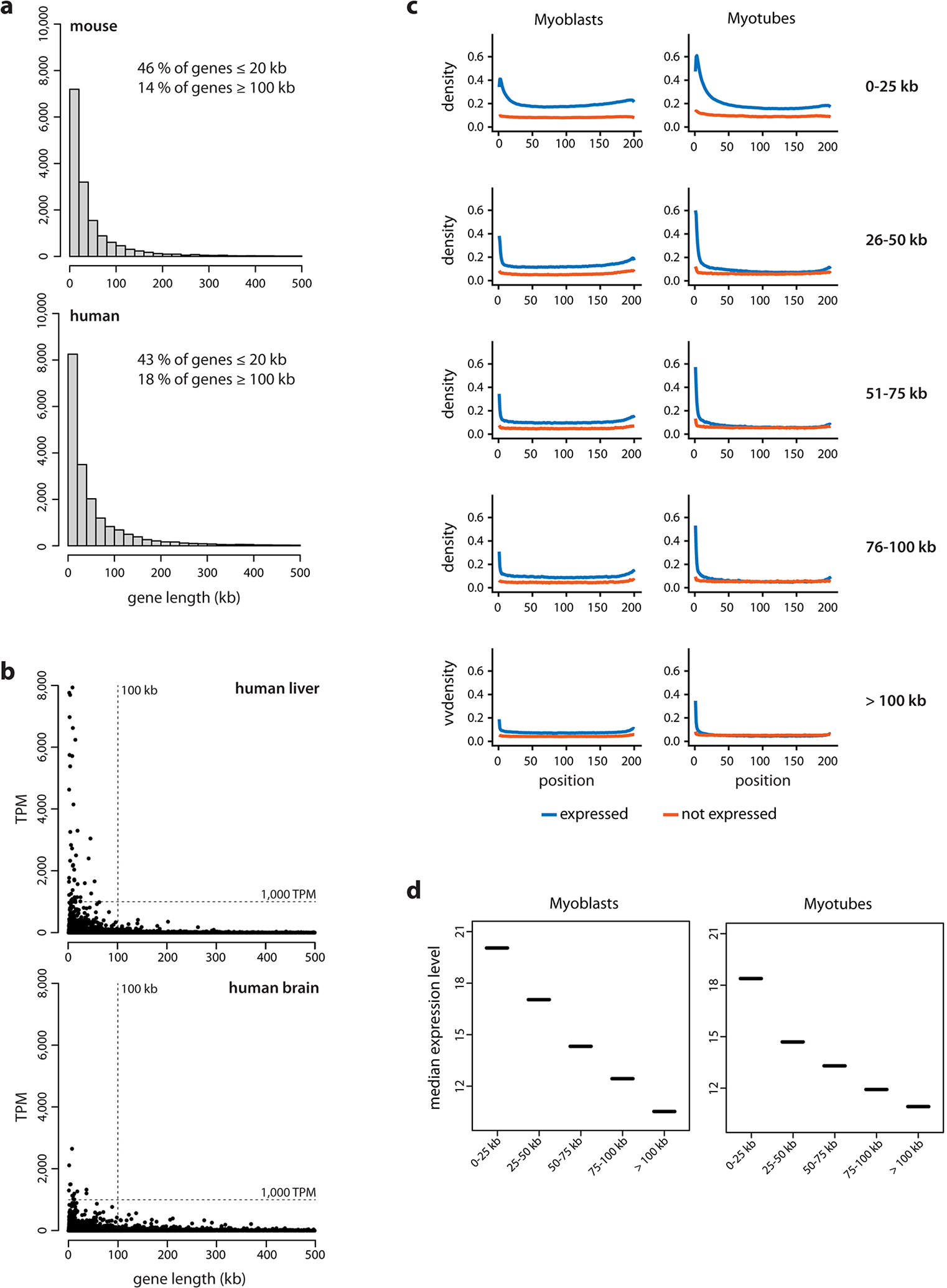
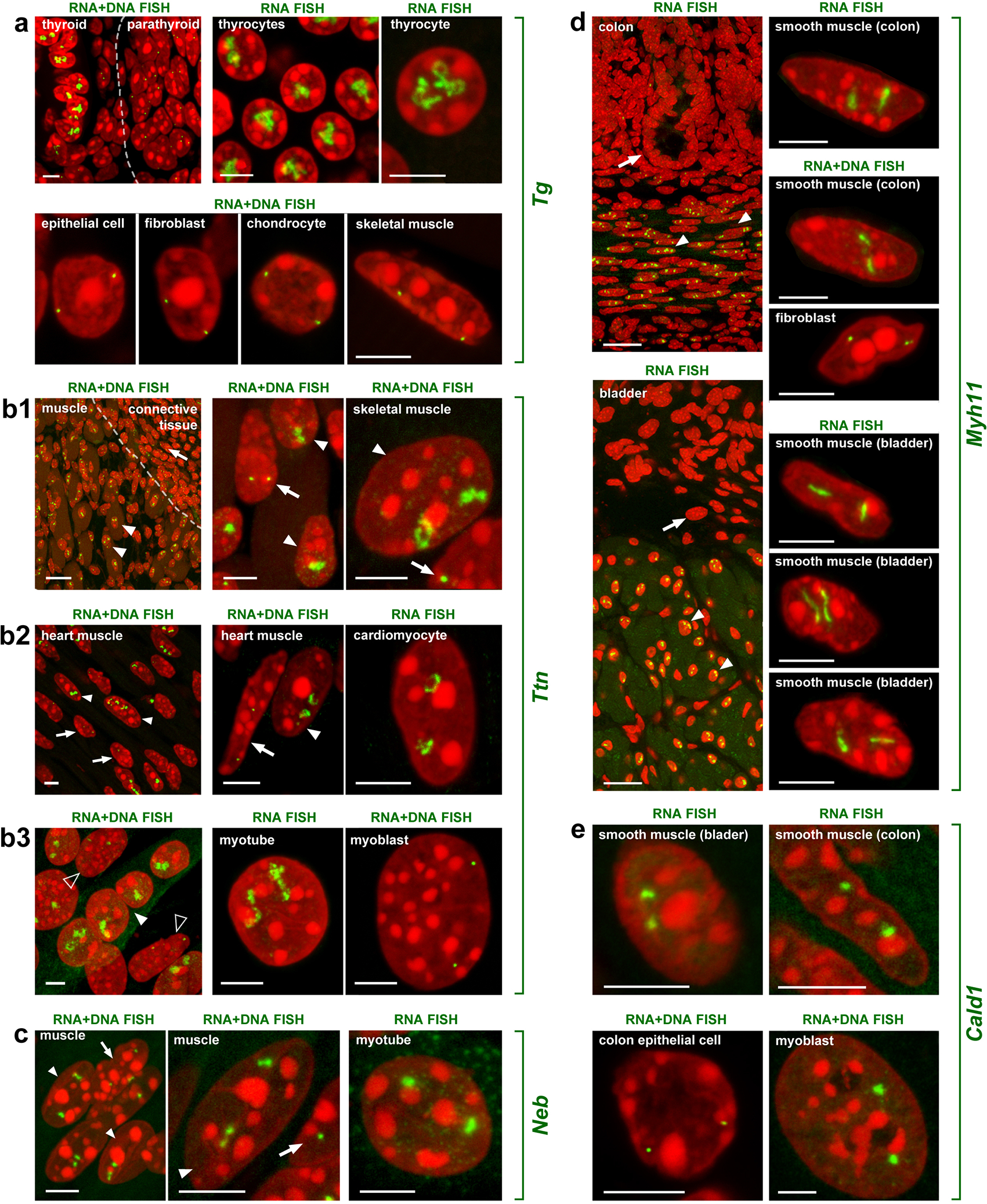
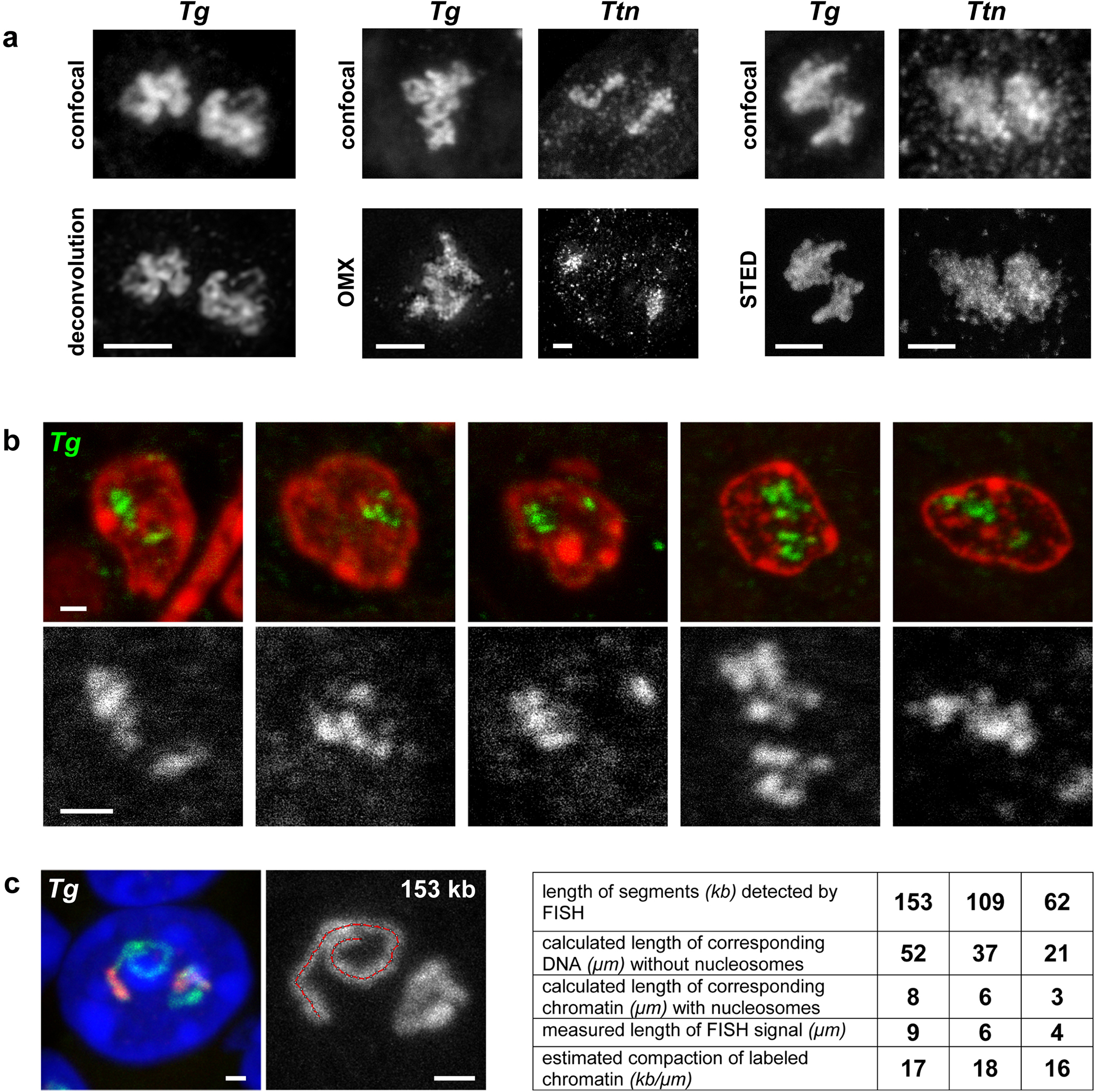
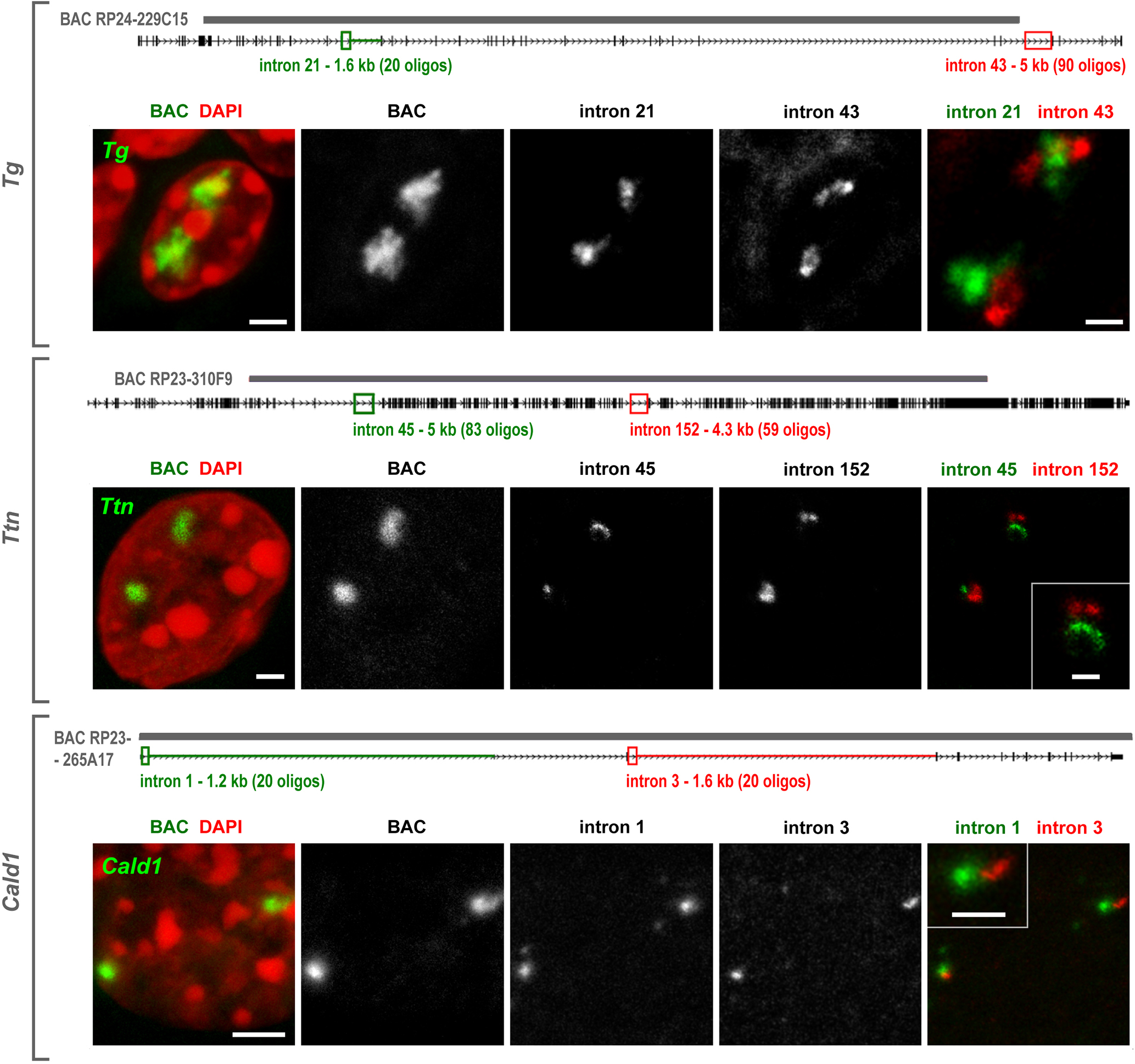
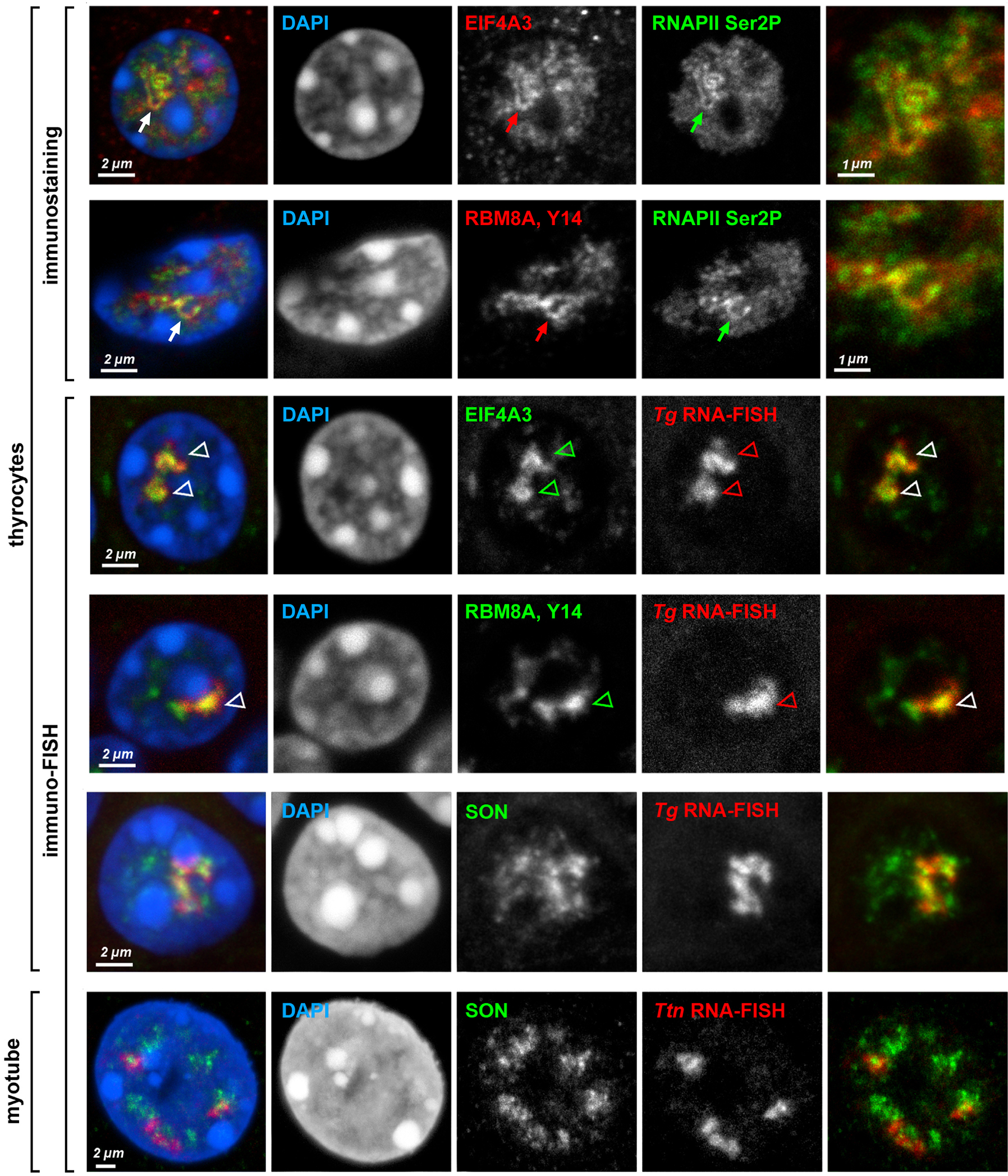
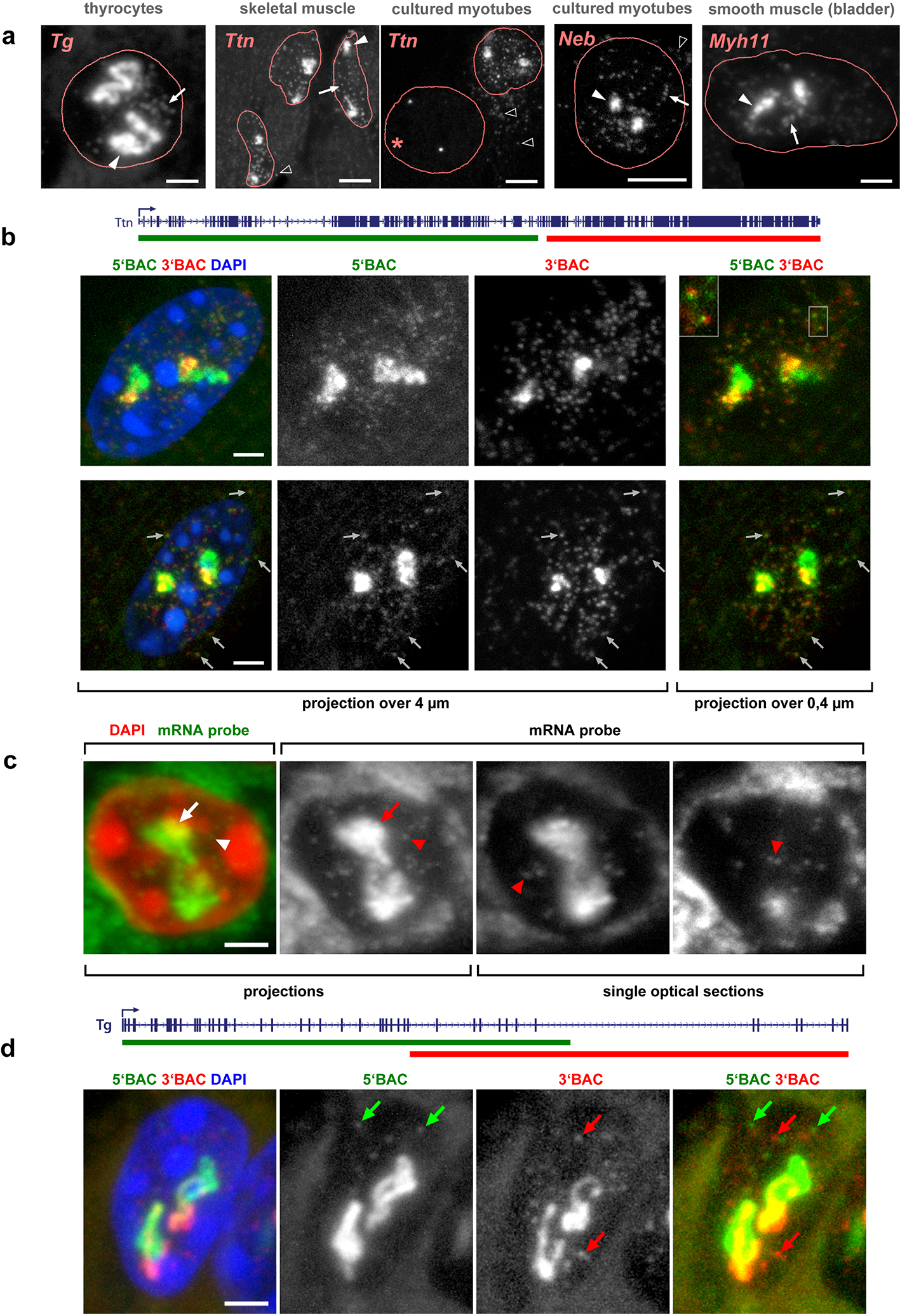
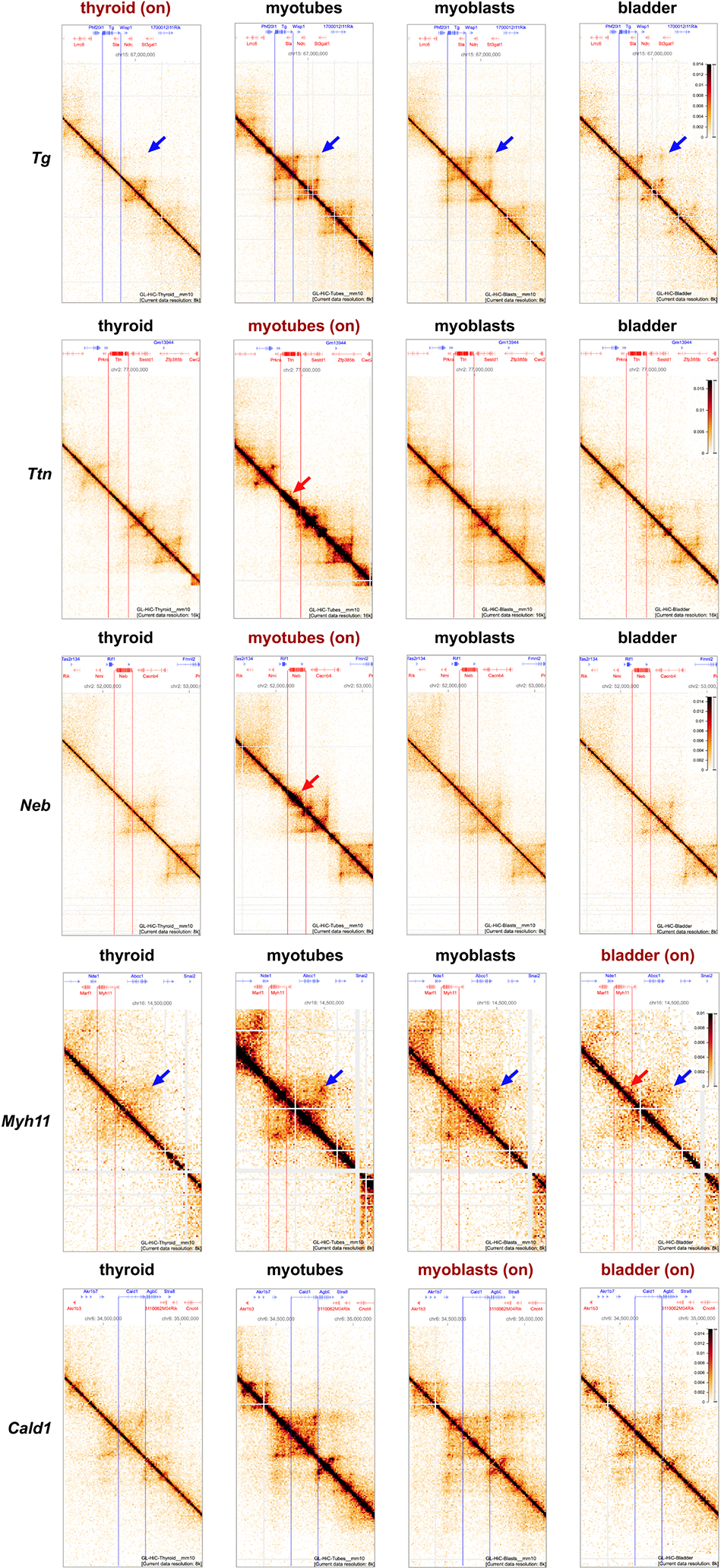
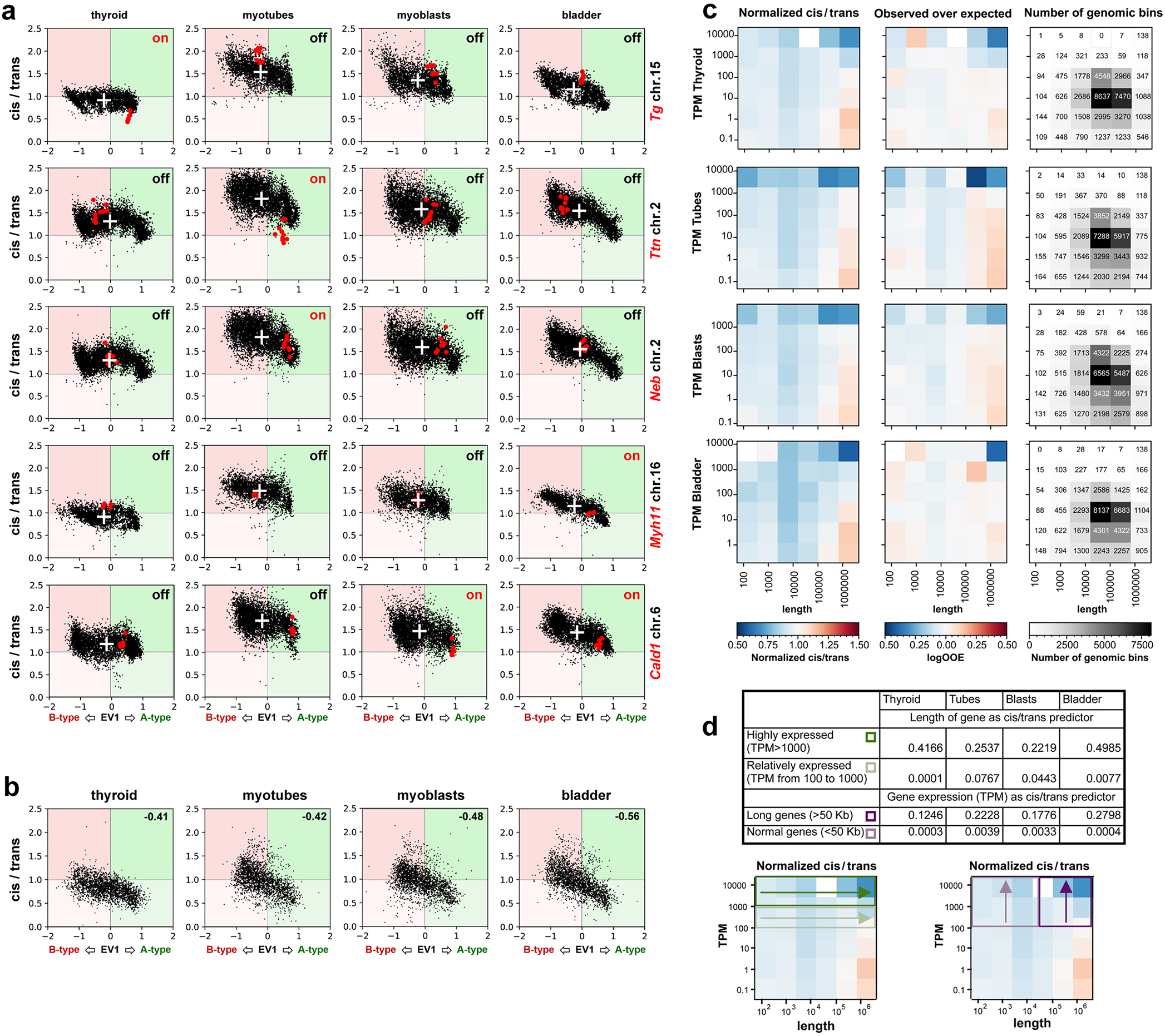
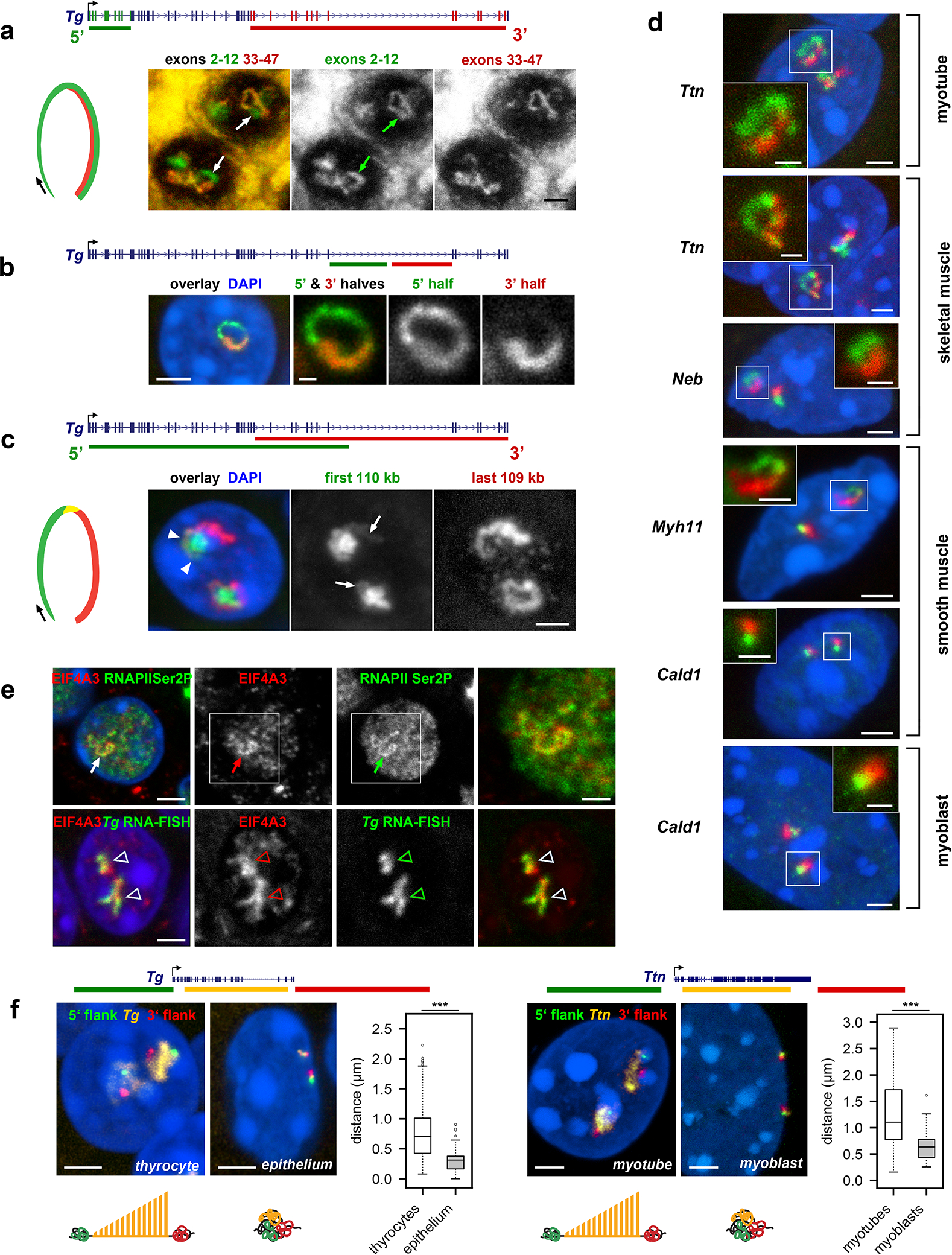
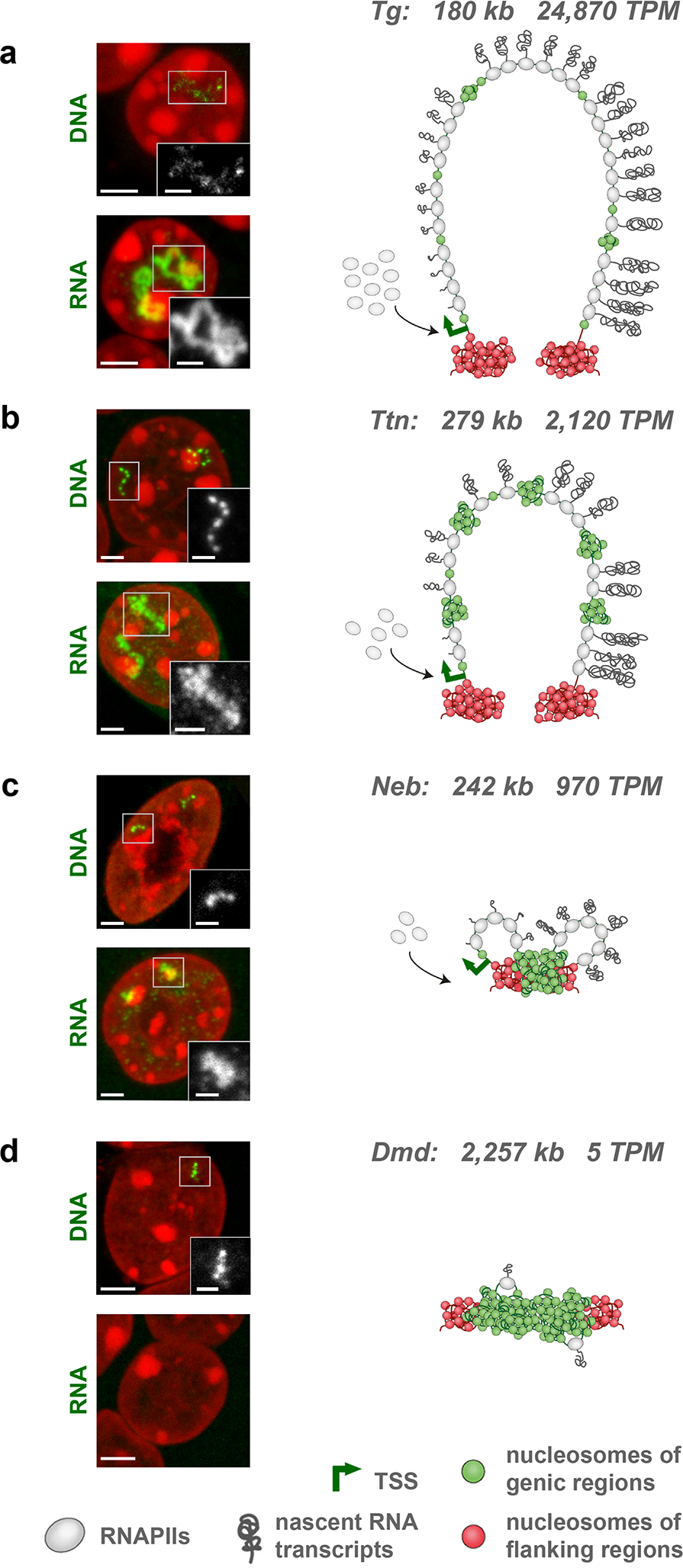
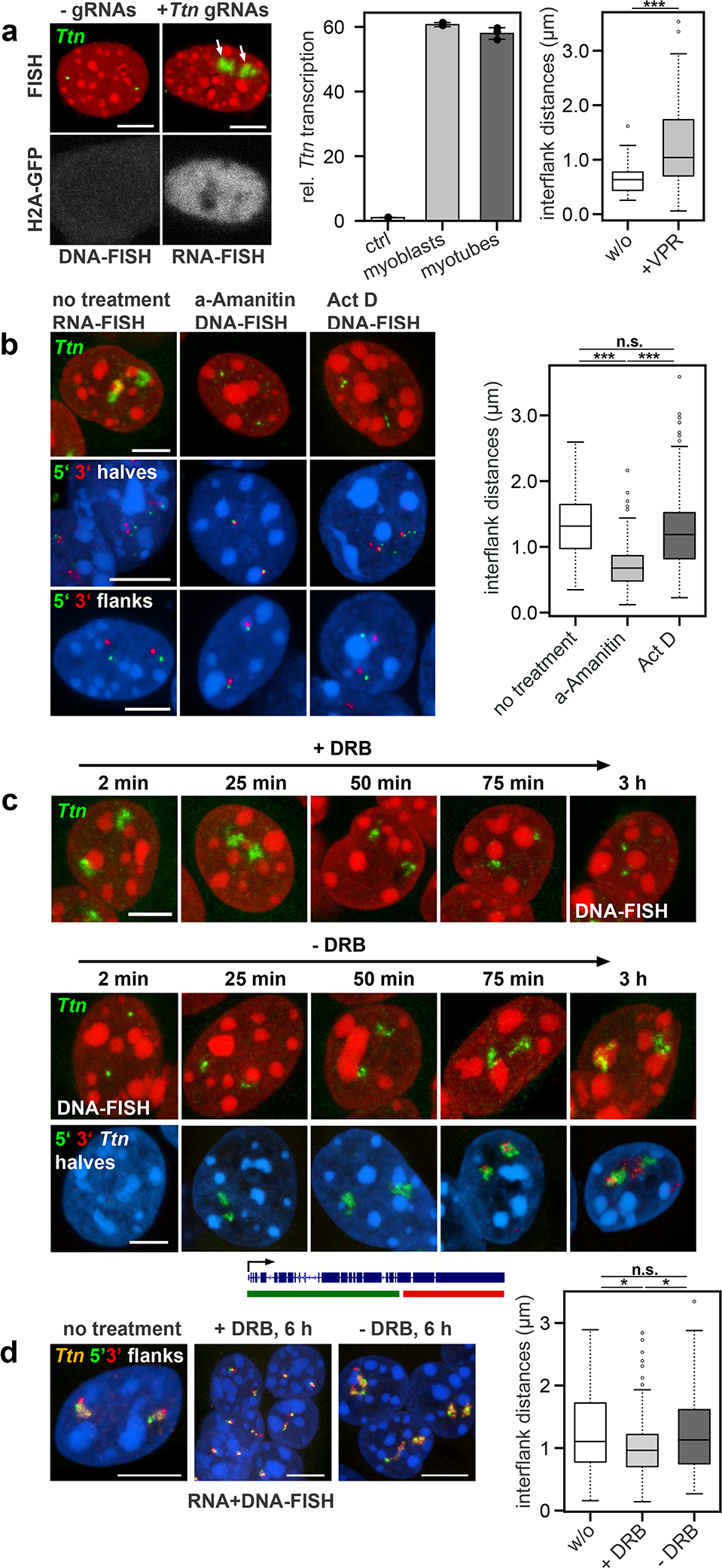
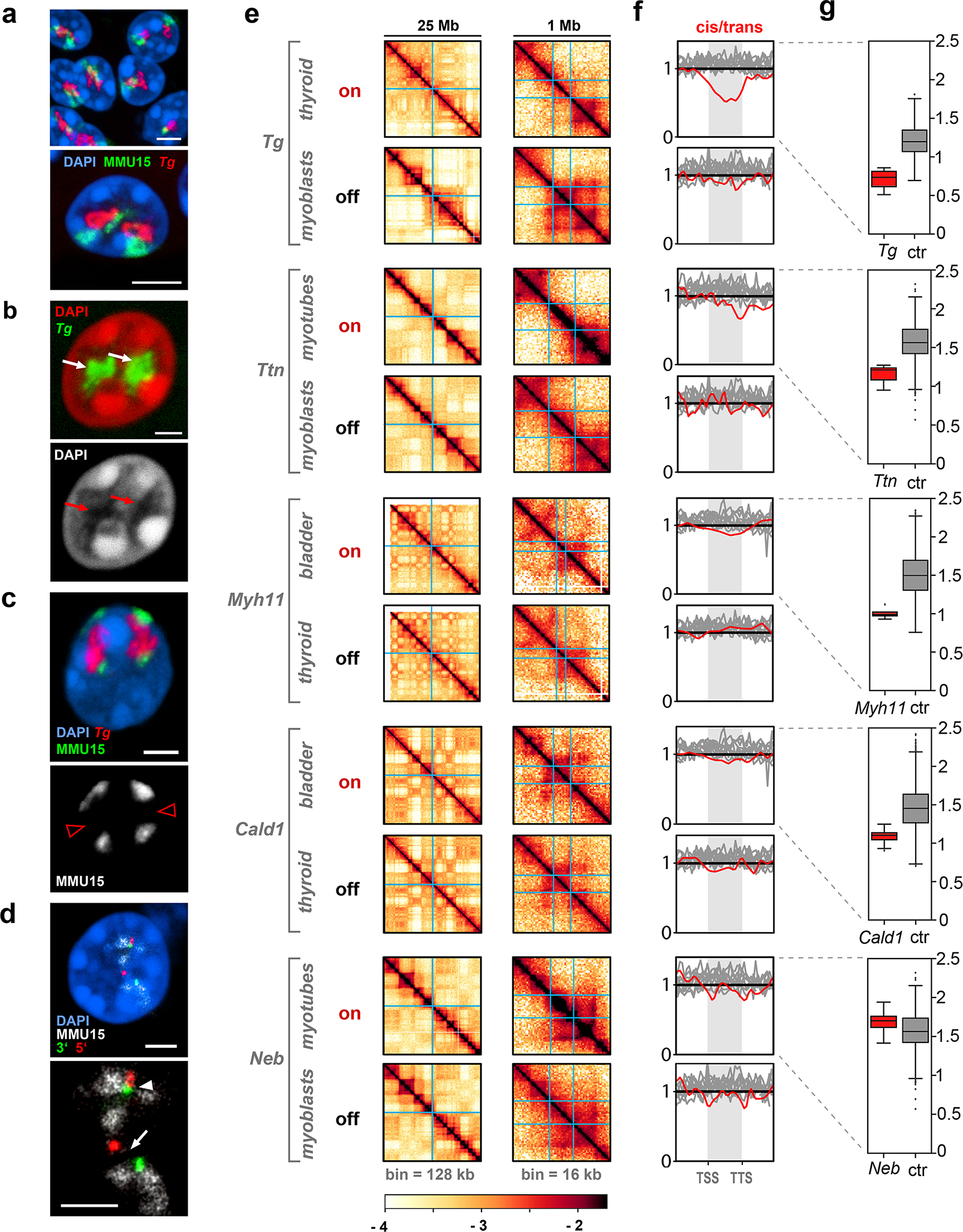
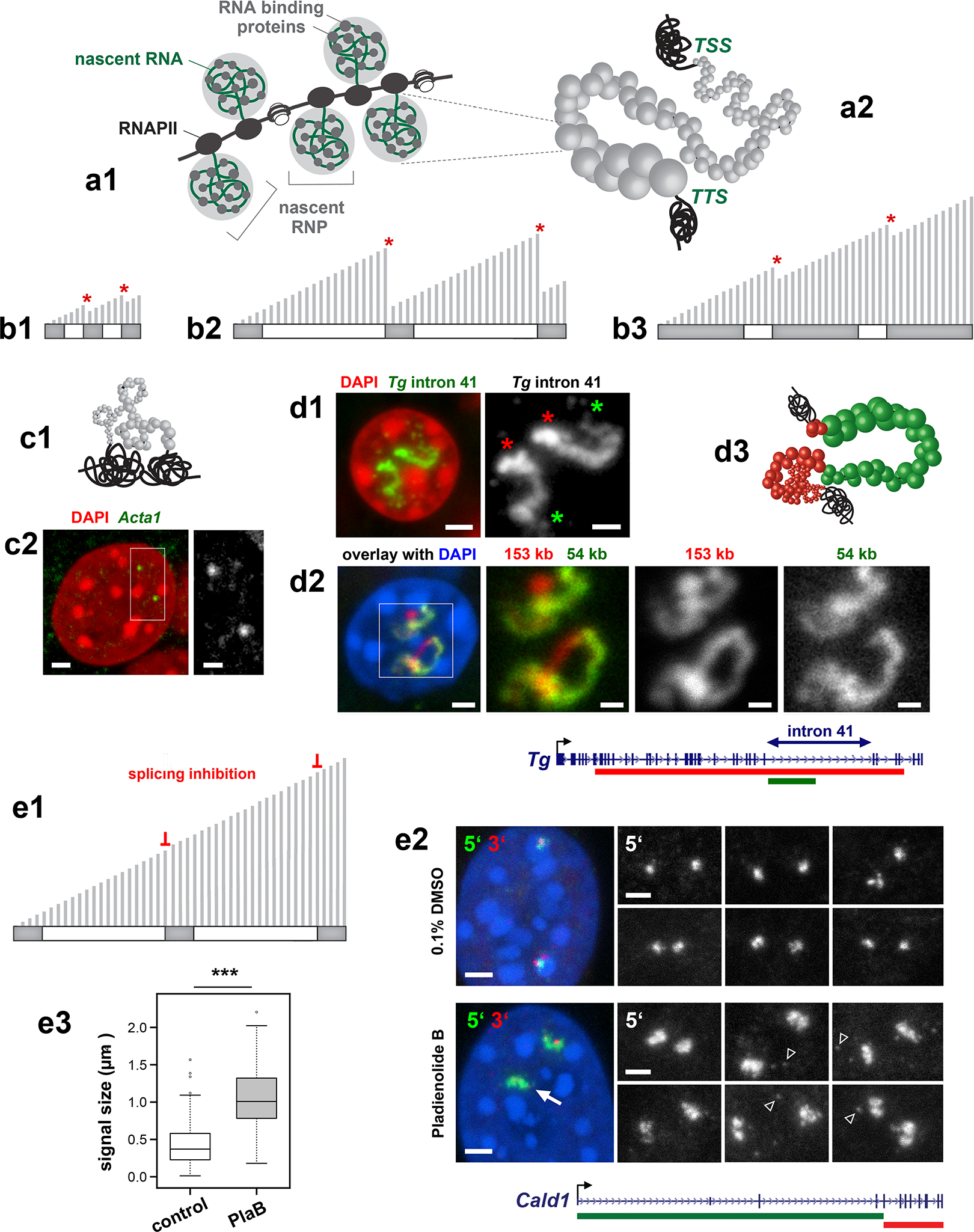
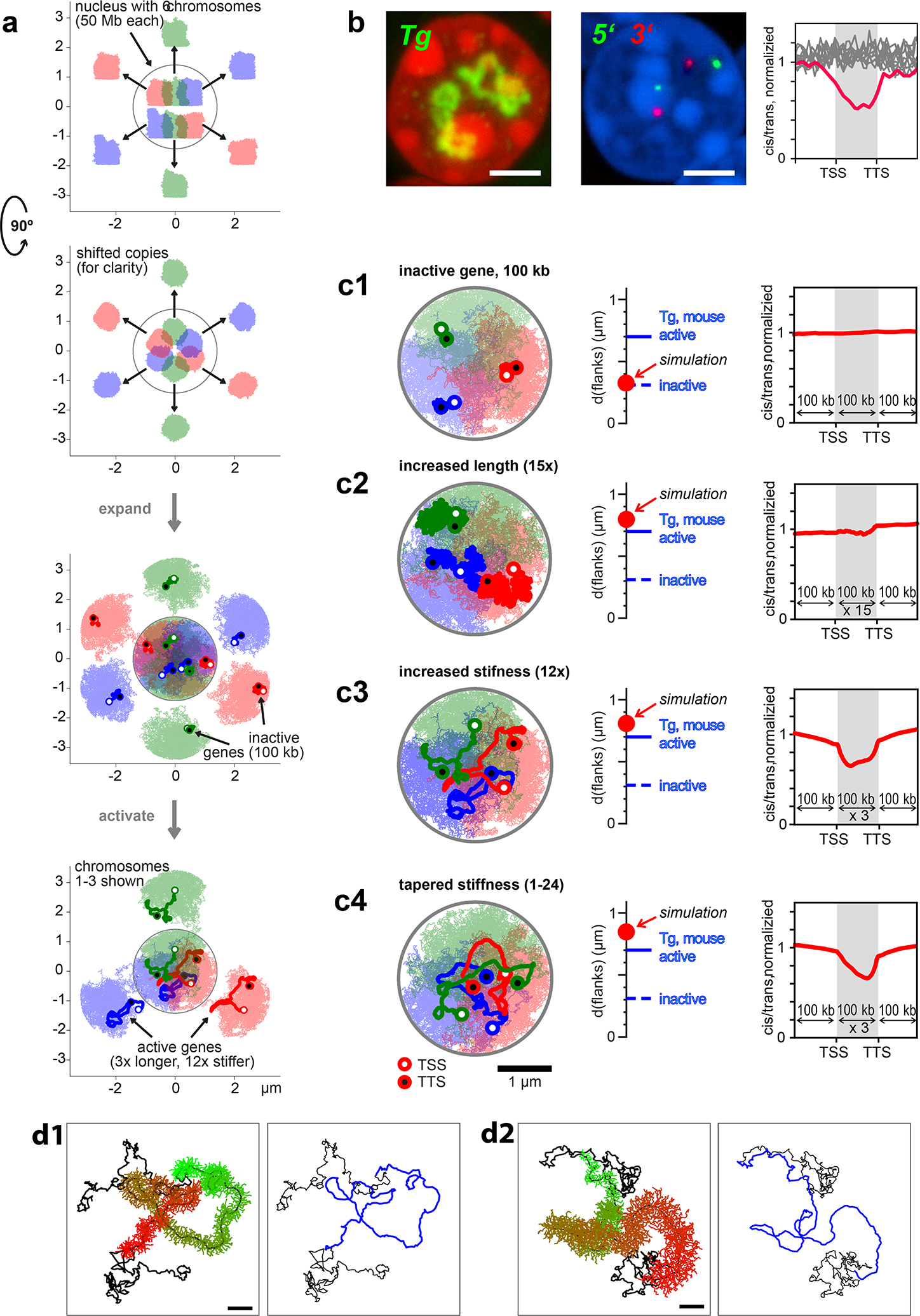
Despite the well-established role of nuclear organization in the regulation of gene expression, little is known about the reverse: how transcription shapes the spatial organization of the genome. Owing to the small sizes of most previously studied genes and the limited resolution of microscopy, the structure and spatial arrangement of a single transcribed gene are still poorly understood. Here we study several long highly expressed genes and demonstrate that they form open-ended transcription loops with polymerases moving along the loops and carrying nascent RNAs. Transcription loops can span across micrometres, resembling lampbrush loops and polytene puffs. The extension and shape of transcription loops suggest their intrinsic stiffness, which we attribute to decoration with multiple voluminous nascent ribonucleoproteins. Our data contradict the model of transcription factories and suggest that although microscopically resolvable transcription loops are specific for long highly expressed genes, the mechanisms underlying their formation could represent a general aspect of eukaryotic transcription.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
COMPETING INTERESTS STATEMENT
The authors declare no competing interests
Figures
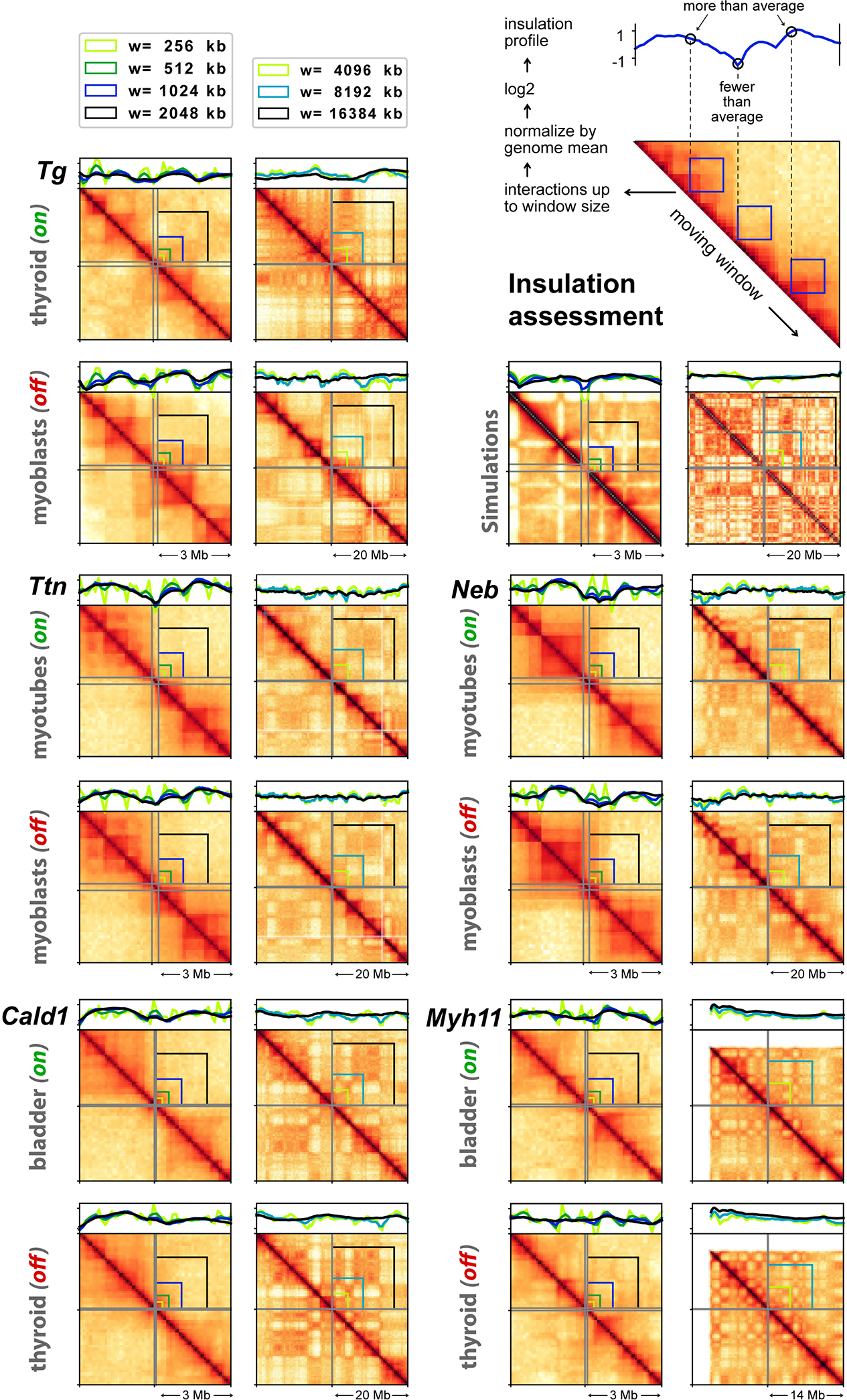
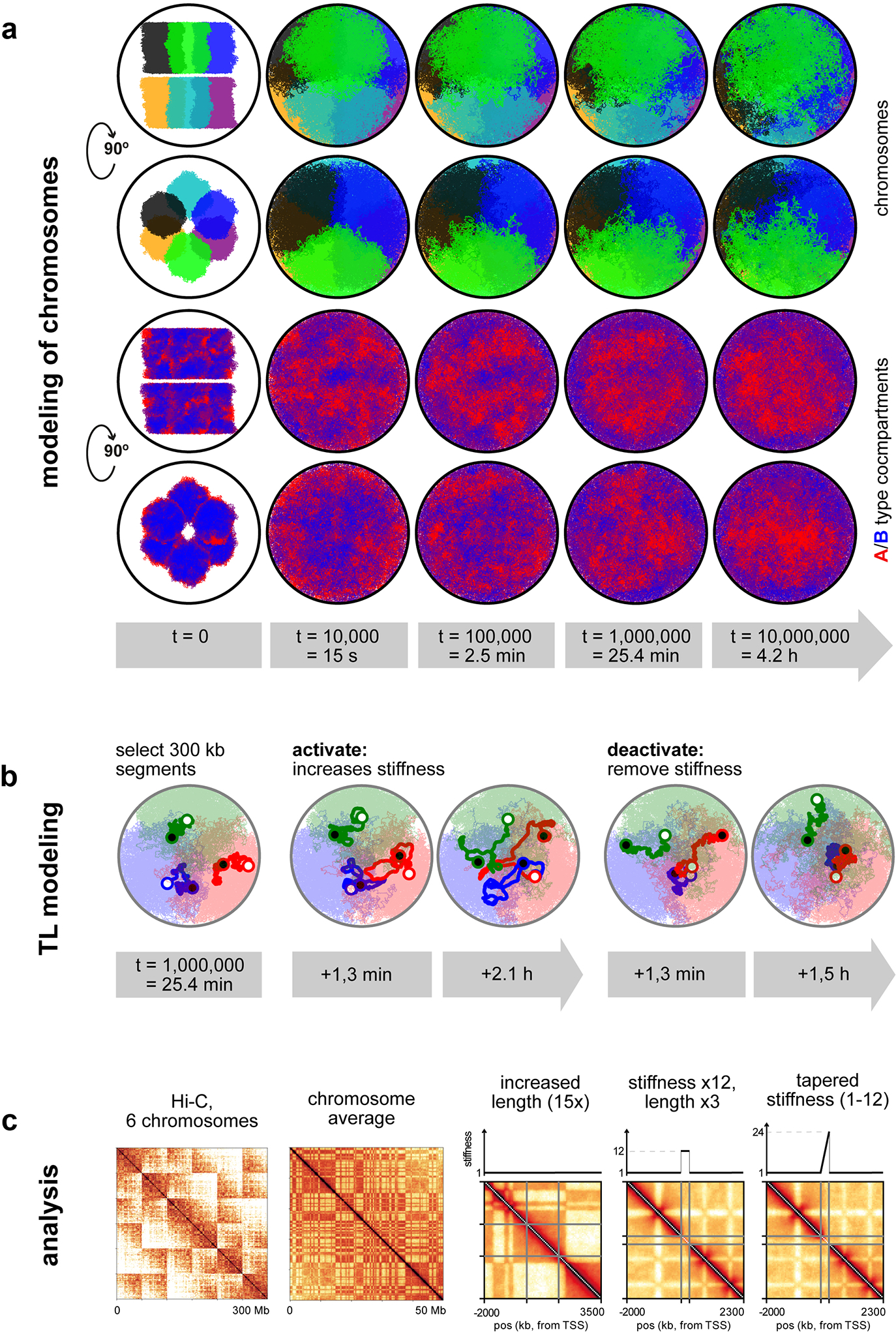
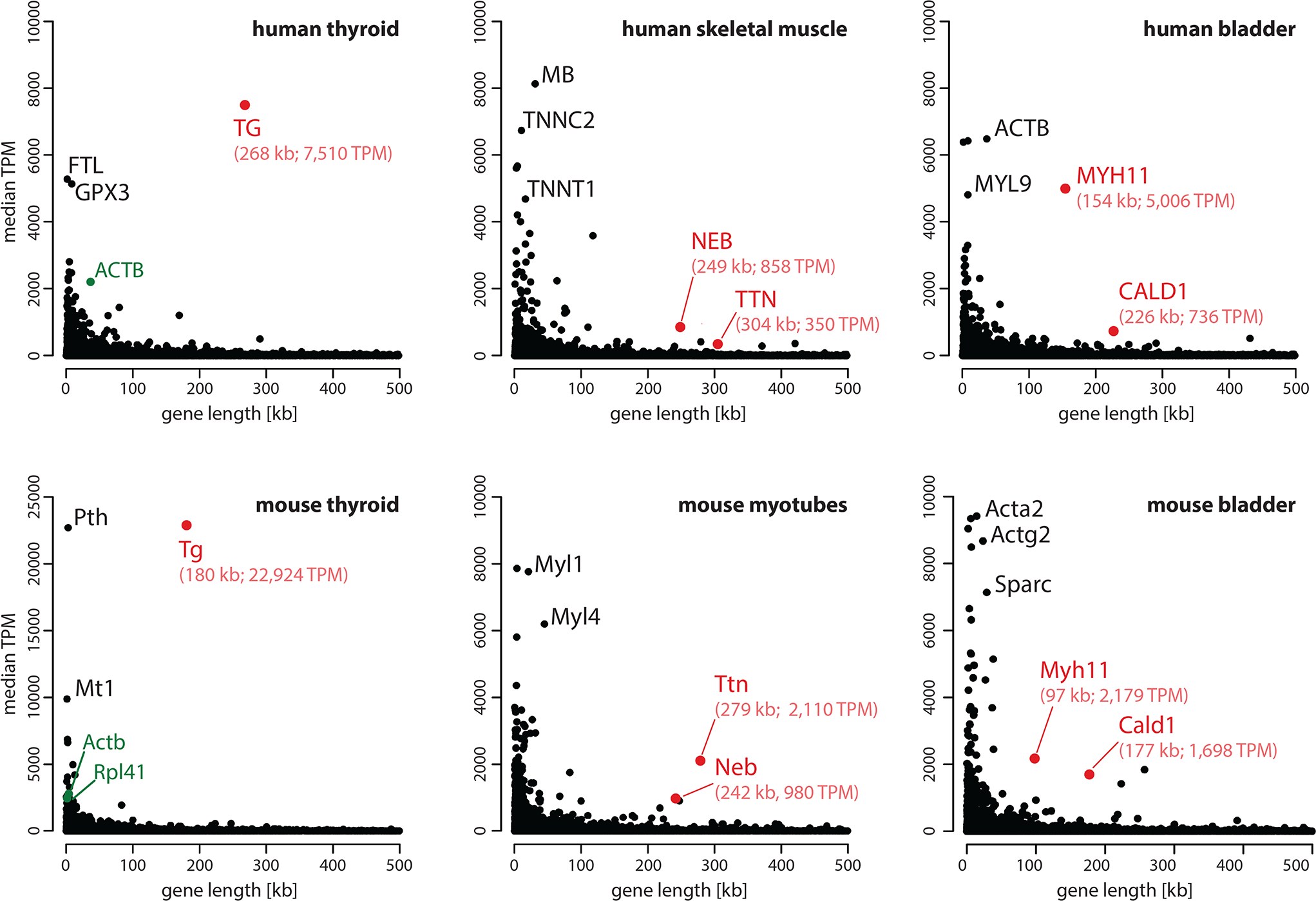
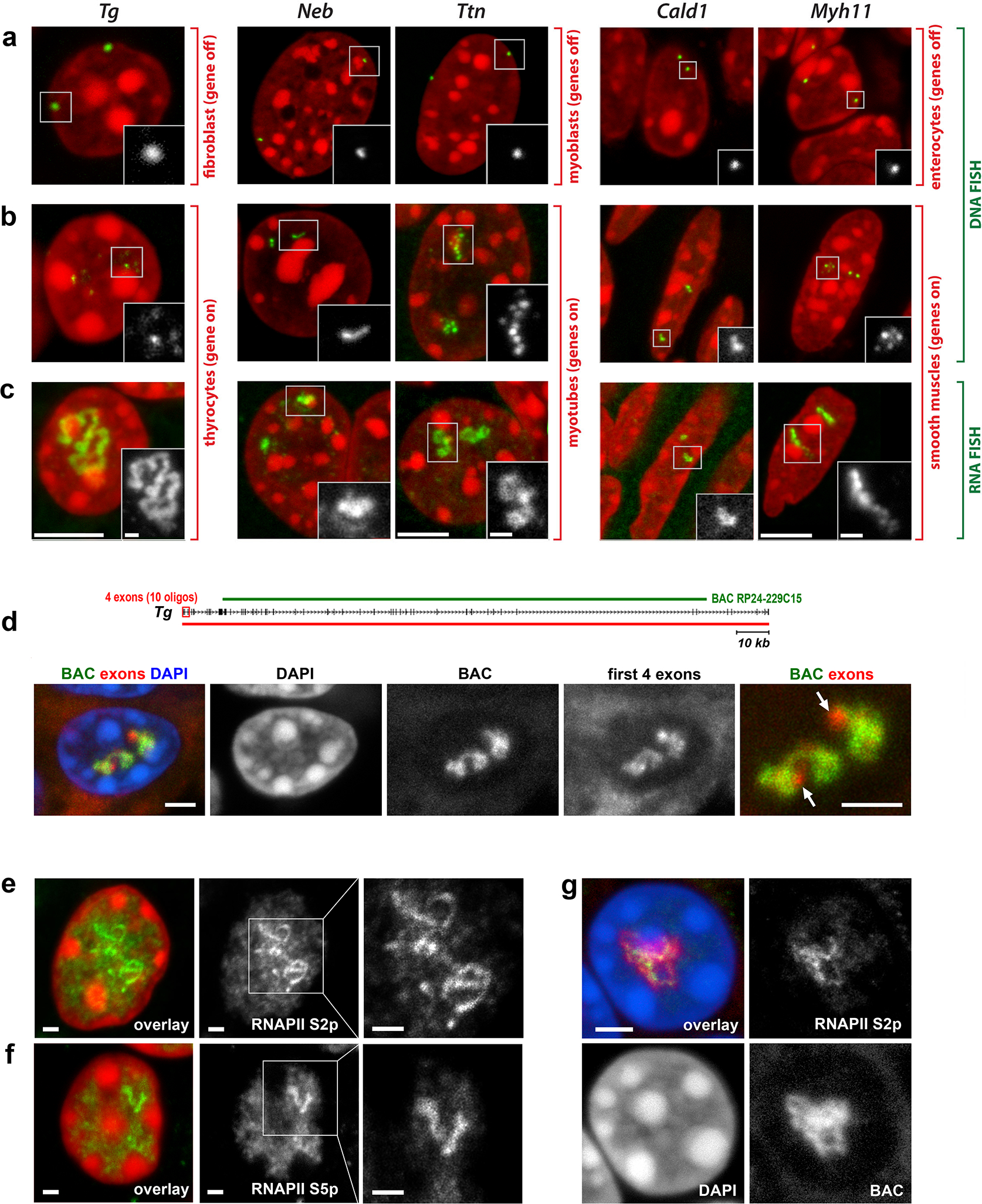
Comment in
-
Transcripts in the loop.Nat Rev Mol Cell Biol. 2022 Apr;23(4):229. doi: 10.1038/s41580-022-00468-9. Nat Rev Mol Cell Biol. 2022. PMID: 35197609 No abstract available.
-
The sight of transcription.Nat Cell Biol. 2022 Mar;24(3):284-285. doi: 10.1038/s41556-022-00865-4. Nat Cell Biol. 2022. PMID: 35256777 No abstract available.
Similar articles
-
Imaging the dynamics of transcription loops in living chromosomes.Chromosoma. 2018 Sep;127(3):361-374. doi: 10.1007/s00412-018-0667-8. Epub 2018 Apr 3. Chromosoma. 2018. PMID: 29610944 Free PMC article.
-
[Chicken lampbrush chromosomes: transcription of tandemly repetitive DNA sequences].Genetika. 2010 Oct;46(10):1329-34. Genetika. 2010. PMID: 21254549 Russian.
-
Small nuclear ribonucleoproteins and heterogeneous nuclear ribonucleoproteins in the amphibian germinal vesicle: loops, spheres, and snurposomes.J Cell Biol. 1991 May;113(3):465-83. doi: 10.1083/jcb.113.3.465. J Cell Biol. 1991. PMID: 1826687 Free PMC article.
-
What role might lampbrush chromosomes play in maternal gene expression?Int J Dev Biol. 1996 Aug;40(4):645-52. Int J Dev Biol. 1996. PMID: 8877436 Review.
-
Lampbrush chromosomes and associated bodies: new insights into principles of nuclear structure and function.Chromosome Res. 2002;10(3):177-200. doi: 10.1023/a:1015227020652. Chromosome Res. 2002. PMID: 12067208 Review.
Cited by
-
Regulation of chromatin microphase separation by binding of protein complexes.Elife. 2023 Jul 12;12:e82983. doi: 10.7554/eLife.82983. Elife. 2023. PMID: 37436818 Free PMC article.
-
Temporal analysis suggests a reciprocal relationship between 3D chromatin structure and transcription.Cell Rep. 2022 Nov 1;41(5):111567. doi: 10.1016/j.celrep.2022.111567. Cell Rep. 2022. PMID: 36323252 Free PMC article.
-
Chromatin alternates between A and B compartments at kilobase scale for subgenic organization.Nat Commun. 2023 Jun 6;14(1):3303. doi: 10.1038/s41467-023-38429-1. Nat Commun. 2023. PMID: 37280210 Free PMC article.
-
Transcription decouples estrogen-dependent changes in enhancer-promoter contact frequencies and spatial proximity.PLoS Genet. 2024 May 23;20(5):e1011277. doi: 10.1371/journal.pgen.1011277. eCollection 2024 May. PLoS Genet. 2024. PMID: 38781242 Free PMC article.
-
The anatomy of transcriptionally active chromatin loops in Drosophila primary spermatocytes using super-resolution microscopy.PLoS Genet. 2023 Mar 3;19(3):e1010654. doi: 10.1371/journal.pgen.1010654. eCollection 2023 Mar. PLoS Genet. 2023. PMID: 36867662 Free PMC article.
References
-
- Andersson R & Sandelin A Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet 21, 71–87 (2020). - PubMed
-
- Cramer P Organization and regulation of gene transcription. Nature 573, 45–54 (2019). - PubMed
-
- Solovei I, Thanisch K & Feodorova Y How to rule the nucleus: divide et impera. Curr Opin Cell Biol 40, 47–59 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
