

Analysis of the Geometric and Electronic Structure of Spin-Coupled Iron-Sulfur Dimers with Broken-Symmetry DFT: Implications for FeMoco
- PMID: 35167749
- PMCID: PMC8908755
- DOI: 10.1021/acs.jctc.1c00753
Analysis of the Geometric and Electronic Structure of Spin-Coupled Iron-Sulfur Dimers with Broken-Symmetry DFT: Implications for FeMoco
Abstract
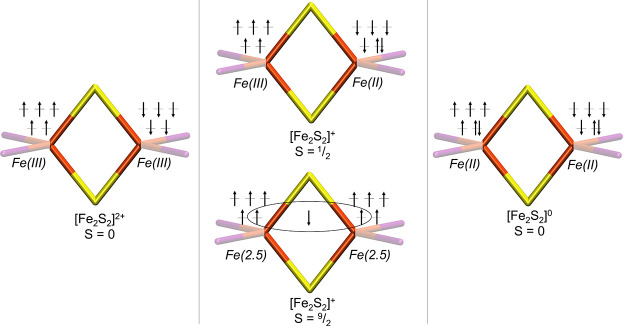
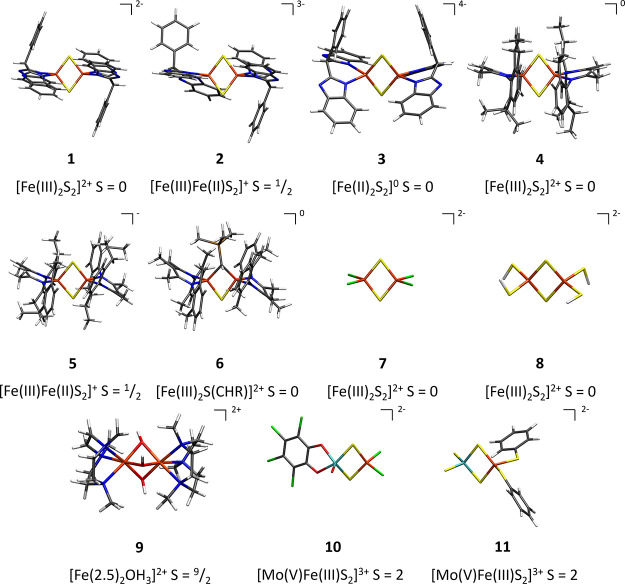
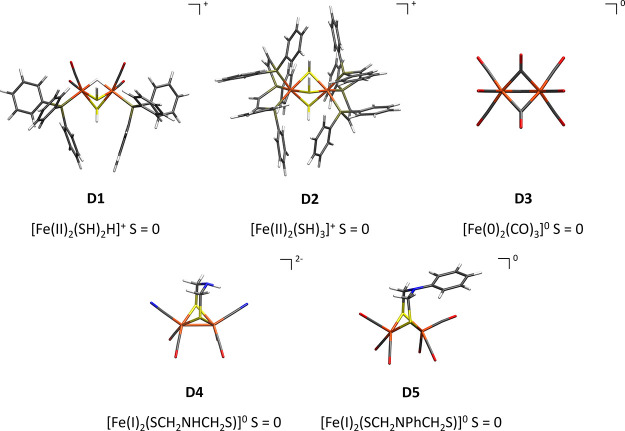
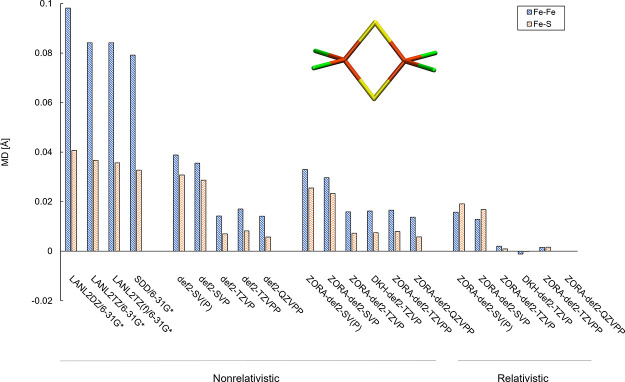
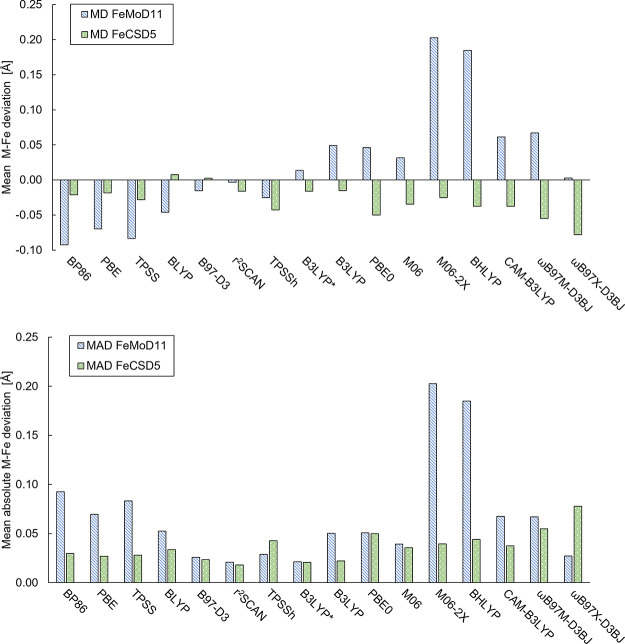
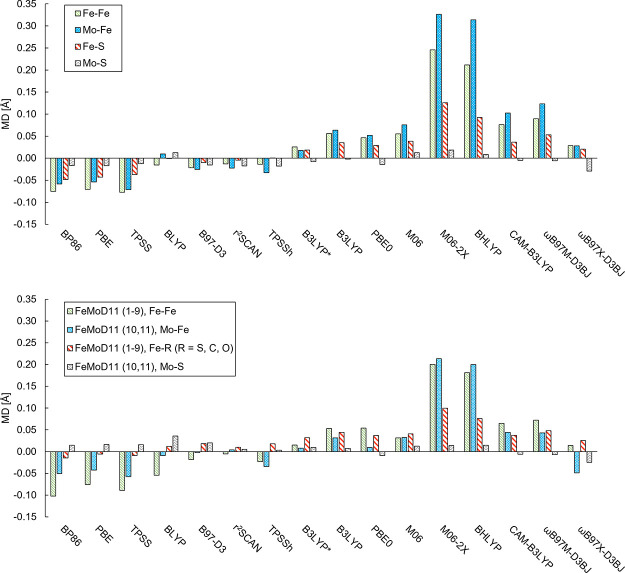
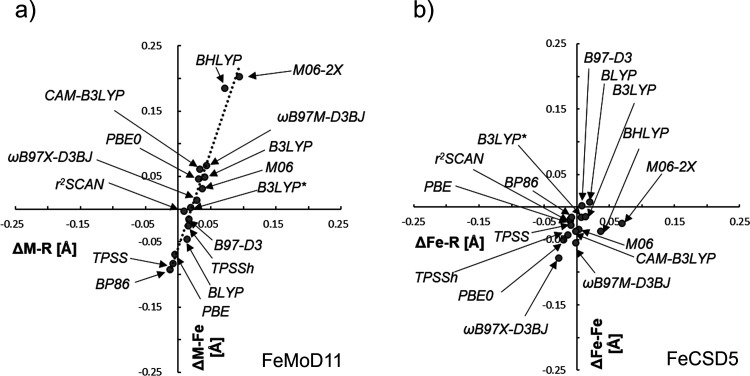
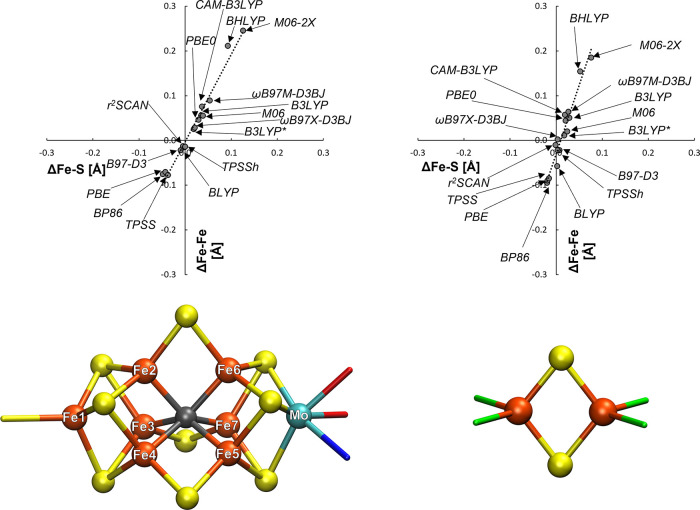
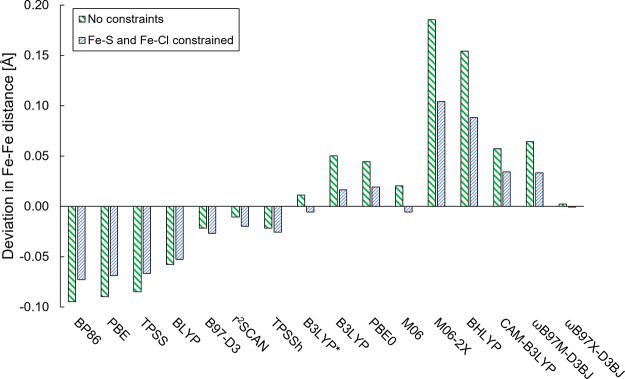
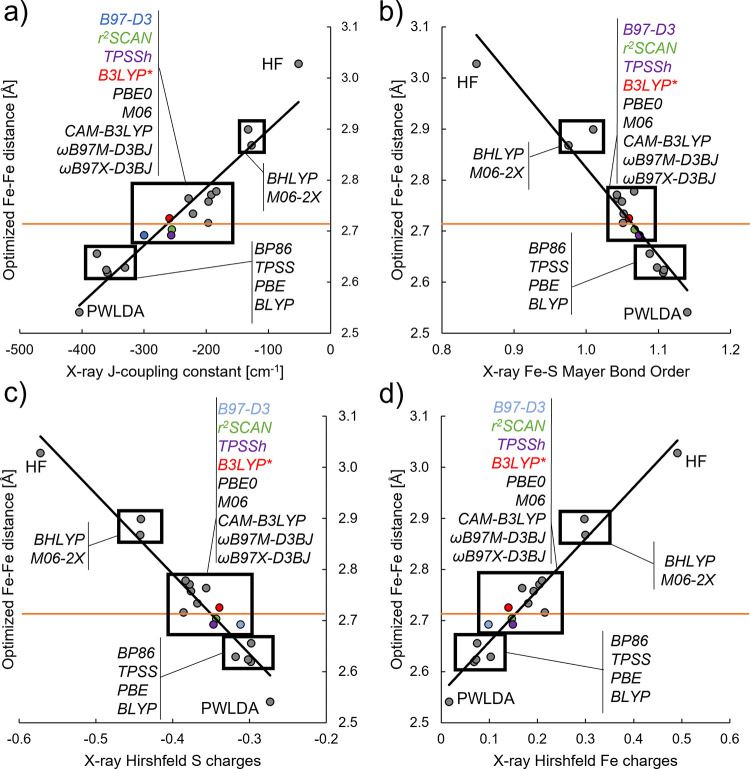
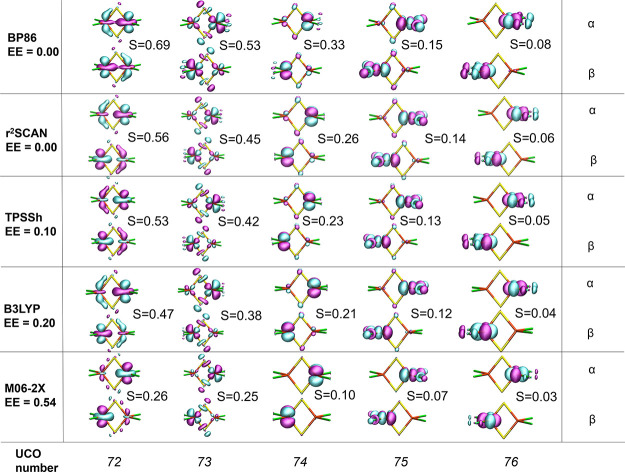
The open-shell electronic structure of iron-sulfur clusters presents considerable challenges to quantum chemistry, with the complex iron-molybdenum cofactor (FeMoco) of nitrogenase representing perhaps the ultimate challenge for either wavefunction or density functional theory. While broken-symmetry density functional theory has seen some success in describing the electronic structure of such cofactors, there is a large exchange-correlation functional dependence in calculations that is not fully understood. In this work, we present a geometric benchmarking test set, FeMoD11, of synthetic spin-coupled Fe-Fe and Mo-Fe dimers, with relevance to the molecular and electronic structure of the Mo-nitrogenase FeMo cofactor. The reference data consists of high-resolution crystal structures of metal dimer compounds in different oxidation states. Multiple density functionals are tested on their ability to reproduce the local geometry, specifically the Fe-Fe/Mo-Fe distance, for both antiferromagnetically coupled and ferromagnetically coupled dimers via the broken-symmetry approach. The metal-metal distance is revealed not only to be highly sensitive to the amount of exact exchange in the functional but also to the specific exchange and correlation functionals. For the antiferromagnetically coupled dimers, the calculated metal-metal distance correlates well with the covalency of the bridging metal-ligand bonds, as revealed via the corresponding orbital analysis, Hirshfeld S/Fe charges, and Fe-S Mayer bond order. Superexchange via bridging ligands is expected to be the dominant interaction in these dimers, and our results suggest that functionals that predict accurate Fe-Fe and Mo-Fe distances describe the overall metal-ligand covalency more accurately and in turn the superexchange of these systems. The best performing density functionals of the 16 tested for the FeMoD11 test set are revealed to be either the nonhybrid functionals r2SCAN and B97-D3 or hybrid functionals with 10-15% exact exchange: TPSSh and B3LYP*. These same four functionals are furthermore found to reproduce the high-resolution X-ray structure of FeMoco well according to quantum mechanics/molecular mechanics (QM/MM) calculations. Almost all nonhybrid functionals systematically underestimate Fe-Fe and Mo-Fe distances (with r2SCAN and B97-D3 being the sole exceptions), while hybrid functionals with >15% exact exchange (including range-separated hybrid functionals) overestimate them. The results overall suggest r2SCAN, B97-D3, TPSSh, and B3LYP* as accurate density functionals for describing the electronic structure of iron-sulfur clusters in general, including the complex FeMoco cluster of nitrogenase.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Understanding the Electronic Structure Basis for N2 Binding to FeMoco: A Systematic Quantum Mechanics/Molecular Mechanics Investigation.Inorg Chem. 2023 Apr 10;62(14):5357-5375. doi: 10.1021/acs.inorgchem.2c03967. Epub 2023 Mar 29. Inorg Chem. 2023. PMID: 36988551 Free PMC article.
-
Quantum Mechanics/Molecular Mechanics Study of Resting-State Vanadium Nitrogenase: Molecular and Electronic Structure of the Iron-Vanadium Cofactor.Inorg Chem. 2020 Aug 17;59(16):11514-11527. doi: 10.1021/acs.inorgchem.0c01320. Epub 2020 Aug 5. Inorg Chem. 2020. PMID: 32799489 Free PMC article.
-
The E3 state of FeMoco: one hydride, two hydrides or dihydrogen?Phys Chem Chem Phys. 2023 Aug 9;25(31):21020-21036. doi: 10.1039/d3cp01106b. Phys Chem Chem Phys. 2023. PMID: 37522223
-
Insight into the Iron-Molybdenum Cofactor of Nitrogenase from Synthetic Iron Complexes with Sulfur, Carbon, and Hydride Ligands.J Am Chem Soc. 2016 Jun 15;138(23):7200-11. doi: 10.1021/jacs.6b00747. Epub 2016 Jun 3. J Am Chem Soc. 2016. PMID: 27171599 Free PMC article. Review.
-
Exploring the potential of natural orbital functionals.Chem Sci. 2024 Oct 10;15(42):17284-91. doi: 10.1039/d4sc05810k. Online ahead of print. Chem Sci. 2024. PMID: 39421199 Free PMC article. Review.
Cited by
-
Benchmarking Density Functional Theory Methods for Metalloenzyme Reactions: The Introduction of the MME55 Set.J Chem Theory Comput. 2023 Nov 28;19(22):8365-8383. doi: 10.1021/acs.jctc.3c00558. Epub 2023 Nov 9. J Chem Theory Comput. 2023. PMID: 37943578 Free PMC article.
-
Correlating Structure with Spectroscopy in Ascorbate Peroxidase Compound II.J Am Chem Soc. 2024 Apr 10;146(14):9640-9656. doi: 10.1021/jacs.3c13169. Epub 2024 Mar 26. J Am Chem Soc. 2024. PMID: 38530124 Free PMC article.
-
Understanding the Electronic Structure Basis for N2 Binding to FeMoco: A Systematic Quantum Mechanics/Molecular Mechanics Investigation.Inorg Chem. 2023 Apr 10;62(14):5357-5375. doi: 10.1021/acs.inorgchem.2c03967. Epub 2023 Mar 29. Inorg Chem. 2023. PMID: 36988551 Free PMC article.
-
Quantum refinement in real and reciprocal space using the Phenix and ORCA software.IUCrJ. 2024 Nov 1;11(Pt 6):921-937. doi: 10.1107/S2052252524008406. IUCrJ. 2024. PMID: 39345101 Free PMC article.
-
Scalar Relativistic All-Electron and Pseudopotential Ab Initio Study of a Minimal Nitrogenase [Fe(SH)4H]- Model Employing Coupled-Cluster and Auxiliary-Field Quantum Monte Carlo Many-Body Methods.J Phys Chem A. 2024 Feb 22;128(7):1358-1374. doi: 10.1021/acs.jpca.3c05808. Epub 2024 Feb 7. J Phys Chem A. 2024. PMID: 38324717 Free PMC article.
References
-
- Lukoyanov D.; Pelmenschikov V.; Maeser N.; Laryukhin M.; Tran C. Y.; Noodleman L.; Dean D. R.; Case D. A.; Seefeldt L. C.; Hoffman B. M. Testing If the Interstitial Atom, X, of the Nitrogenase Molybdenum-Iron Cofactor Is N or C: ENDOR, ESEEM, and DFT Studies of the S = 3/2 Resting State in Multiple Environments. Inorg. Chem. 2007, 46, 11437–11449. 10.1021/ic7018814. - DOI - PubMed
-
- Davydov R.; Khadka N.; Yang Z.; Fielding A. J.; Lukoyanov D.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Exploring Electron/Proton Transfer and Conformational Changes in the Nitrogenase MoFe Protein and FeMo-cofactor Through Cryoreduction/EPR Measurements. Isr. J. Chem. 2016, 56, 841–851. 10.1002/ijch.201600026. - DOI - PMC - PubMed
-
- Hoeke V.; Tociu L.; Case D. A.; Seefeldt L. C.; Raugei S.; Hoffman B. M. High Resolution ENDOR Spectroscopy Combined with Quantum Chemical Calculations Reveals the Structure of the Nitrogenase Janus Intermediate E4(4H). J. Am. Chem. Soc. 2019, 141, 11984–11996. 10.1021/jacs.9b04474. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
