

Curated variation benchmarks for challenging medically relevant autosomal genes
- PMID: 35132260
- PMCID: PMC9117392
- DOI: 10.1038/s41587-021-01158-1
Curated variation benchmarks for challenging medically relevant autosomal genes
Abstract
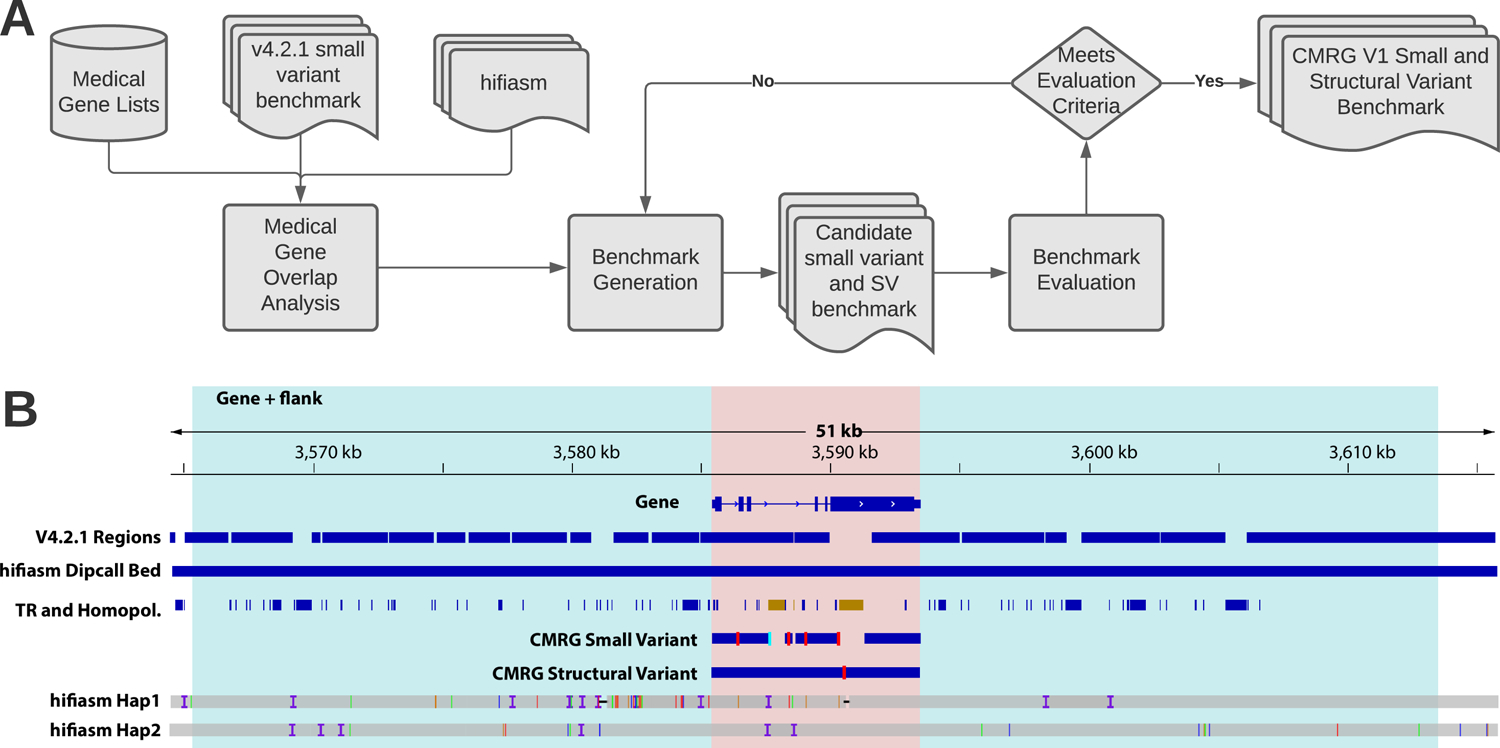
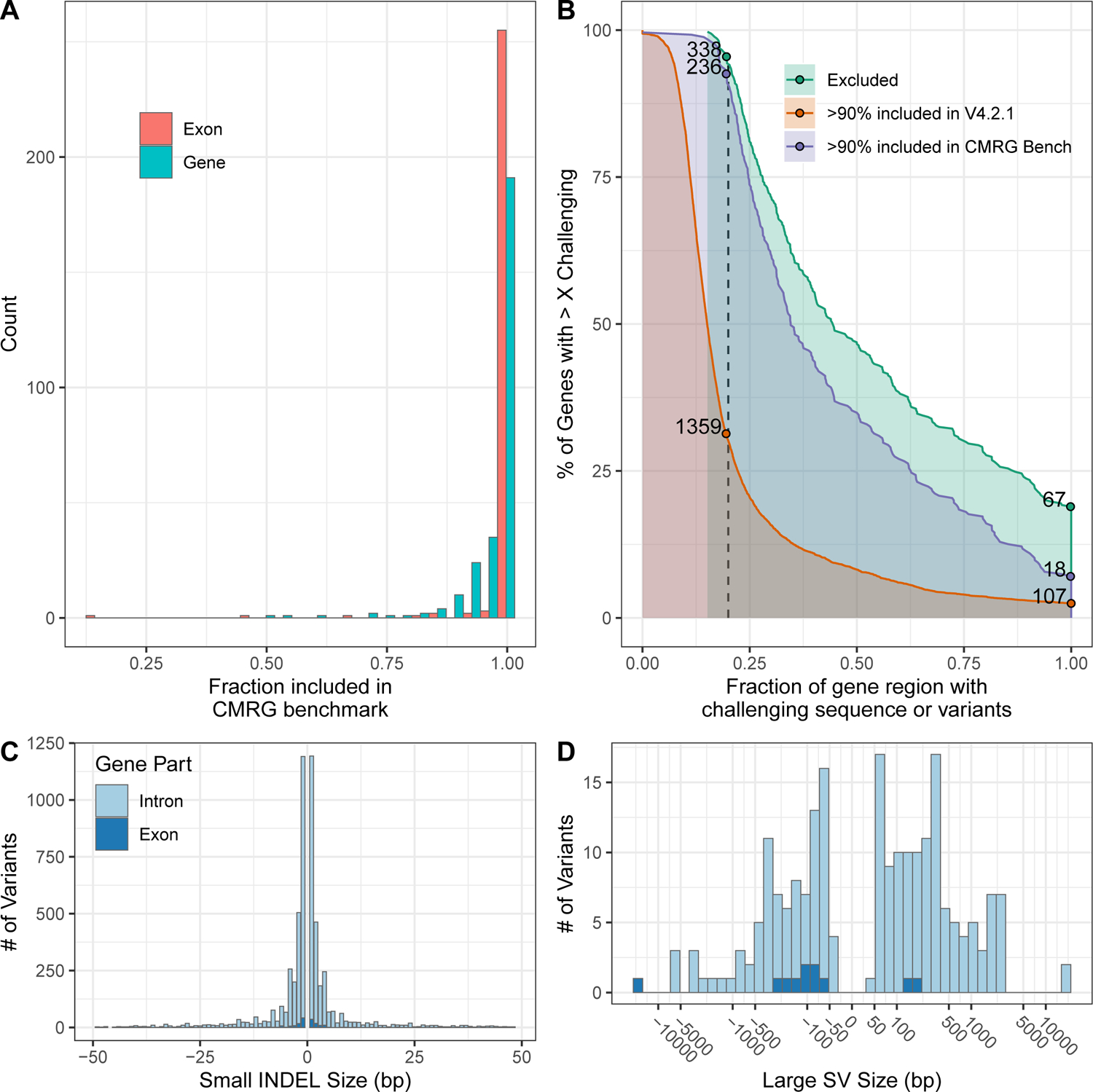
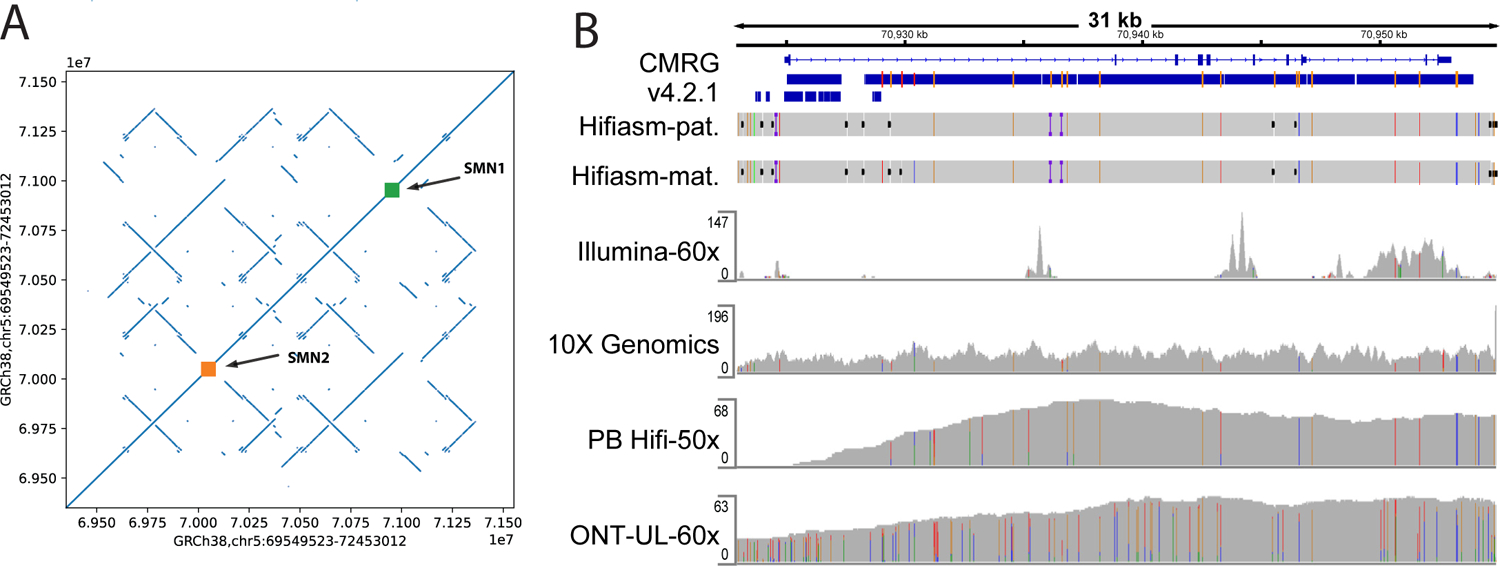
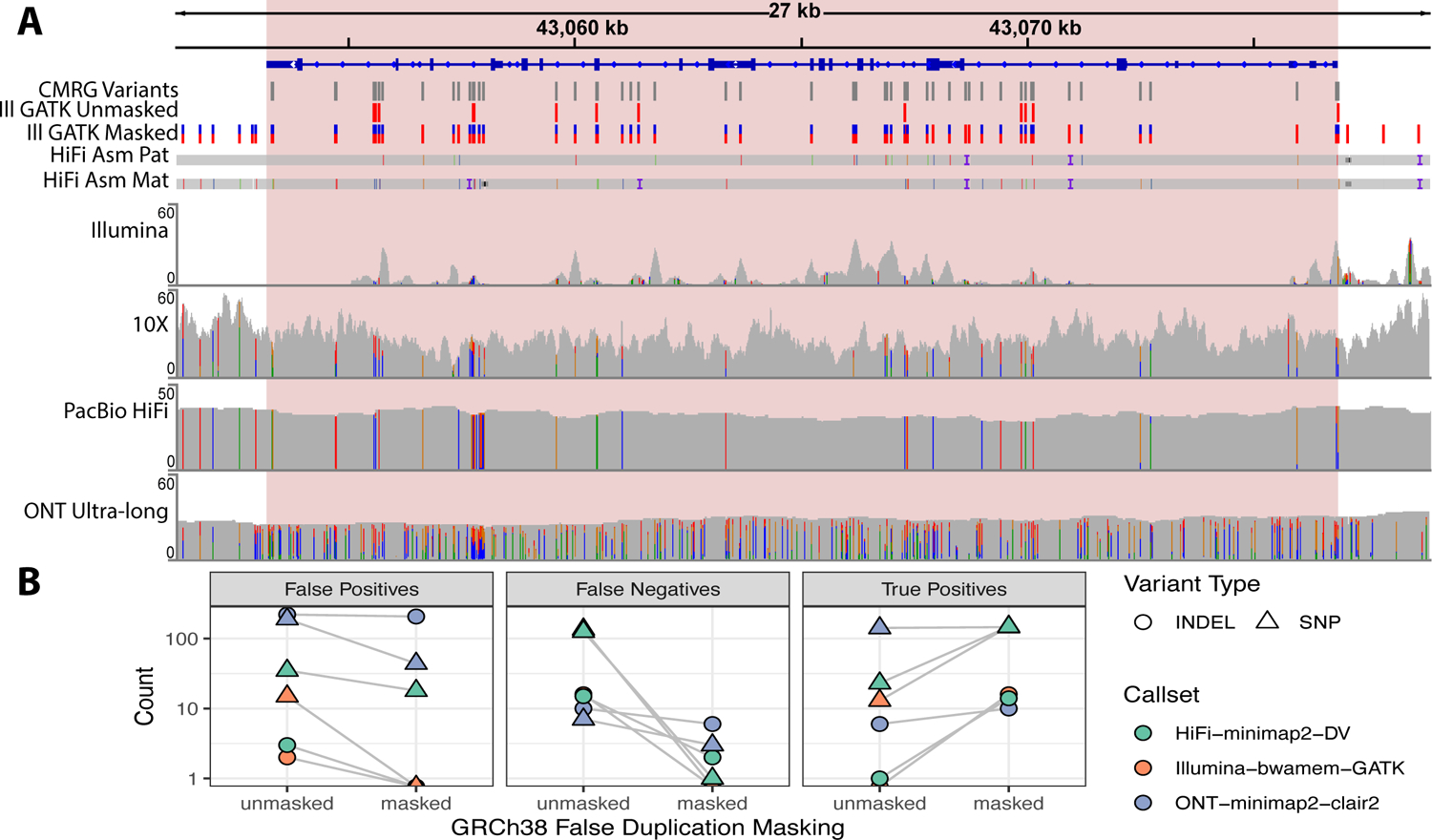
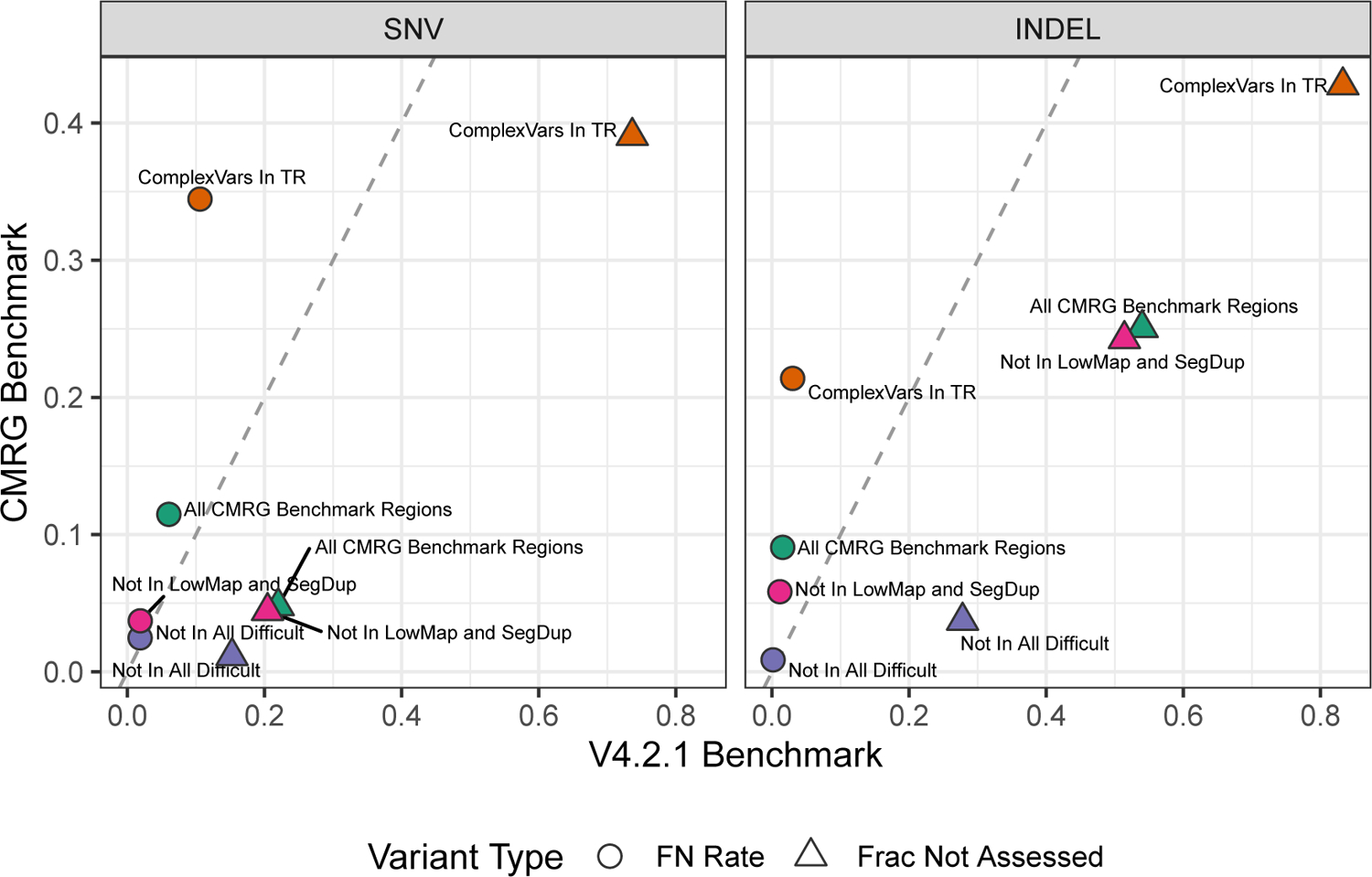
The repetitive nature and complexity of some medically relevant genes poses a challenge for their accurate analysis in a clinical setting. The Genome in a Bottle Consortium has provided variant benchmark sets, but these exclude nearly 400 medically relevant genes due to their repetitiveness or polymorphic complexity. Here, we characterize 273 of these 395 challenging autosomal genes using a haplotype-resolved whole-genome assembly. This curated benchmark reports over 17,000 single-nucleotide variations, 3,600 insertions and deletions and 200 structural variations each for human genome reference GRCh37 and GRCh38 across HG002. We show that false duplications in either GRCh37 or GRCh38 result in reference-specific, missed variants for short- and long-read technologies in medically relevant genes, including CBS, CRYAA and KCNE1. When masking these false duplications, variant recall can improve from 8% to 100%. Forming benchmarks from a haplotype-resolved whole-genome assembly may become a prototype for future benchmarks covering the whole genome.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing Interests
AMW and WJR are employees and shareholders of Pacific Biosciences. AF and CSC are employees and shareholders of DNAnexus. SMES is an employee of Roche. JL is an employee of Bionano Genomics. SEL was an employee of Invitae. FJS has sponsored travel from Pacific Biosciences and Oxford Nanopore. The remaining authors declare no competing interests.
Figures
Similar articles
-
Benchmarking challenging small variants with linked and long reads.Cell Genom. 2022 May;2(5):100128. doi: 10.1016/j.xgen.2022.100128. Cell Genom. 2022. PMID: 36452119 Free PMC article.
-
Clinical Validation of Genome Reference Consortium Human Build 38 in a Laboratory Utilizing Next-Generation Sequencing Technologies.Clin Chem. 2022 Sep 1;68(9):1177-1183. doi: 10.1093/clinchem/hvac113. Clin Chem. 2022. PMID: 35869940
-
Unveiling novel genetic variants in 370 challenging medically relevant genes using the long read sequencing data of 41 samples from 19 global populations.Mol Genet Genomics. 2024 Jul 7;299(1):65. doi: 10.1007/s00438-024-02158-x. Mol Genet Genomics. 2024. PMID: 38972030
-
Haplotyping-Assisted Diploid Assembly and Variant Detection with Linked Reads.Methods Mol Biol. 2023;2590:161-182. doi: 10.1007/978-1-0716-2819-5_11. Methods Mol Biol. 2023. PMID: 36335499 Review.
-
Variant calling and benchmarking in an era of complete human genome sequences.Nat Rev Genet. 2023 Jul;24(7):464-483. doi: 10.1038/s41576-023-00590-0. Epub 2023 Apr 14. Nat Rev Genet. 2023. PMID: 37059810 Review.
Cited by
-
Semi-automated assembly of high-quality diploid human reference genomes.Nature. 2022 Nov;611(7936):519-531. doi: 10.1038/s41586-022-05325-5. Epub 2022 Oct 19. Nature. 2022. PMID: 36261518 Free PMC article.
-
Resolving intra-repeat variation in medically relevant VNTRs from short-read sequencing data using the cardiovascular risk gene LPA as a model.Genome Biol. 2024 Jun 26;25(1):167. doi: 10.1186/s13059-024-03316-5. Genome Biol. 2024. PMID: 38926899 Free PMC article.
-
Pangenomic genotyping with the marker array.Algorithms Mol Biol. 2023 May 5;18(1):2. doi: 10.1186/s13015-023-00225-3. Algorithms Mol Biol. 2023. PMID: 37147657 Free PMC article.
-
A multilocus approach for accurate variant calling in low-copy repeats using whole-genome sequencing.Bioinformatics. 2023 Jun 30;39(39 Suppl 1):i279-i287. doi: 10.1093/bioinformatics/btad268. Bioinformatics. 2023. PMID: 37387146 Free PMC article.
-
Pangenome graph construction from genome alignments with Minigraph-Cactus.Nat Biotechnol. 2024 Apr;42(4):663-673. doi: 10.1038/s41587-023-01793-w. Epub 2023 May 10. Nat Biotechnol. 2024. PMID: 37165083 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
- R01 AI151059/AI/NIAID NIH HHS/United States
- R01 HG010040/HG/NHGRI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- 9999-NIST/ImNIST/Intramural NIST DOC/United States
- R01 AG068331/AG/NIA NIH HHS/United States
- UM1 HG008898/HG/NHGRI NIH HHS/United States
- R01 CA249054/CA/NCI NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- U01 DA053941/DA/NIDA NIH HHS/United States
- R01 MH117406/MH/NIMH NIH HHS/United States
- P01 CA214274/CA/NCI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- L30 HG009212/HG/NHGRI NIH HHS/United States
- R35 GM138636/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
