

Kinin B1 Receptor Mediates Renal Injury and Remodeling in Hypertension
- PMID: 35118089
- PMCID: PMC8804098
- DOI: 10.3389/fmed.2021.780834
Kinin B1 Receptor Mediates Renal Injury and Remodeling in Hypertension
Abstract
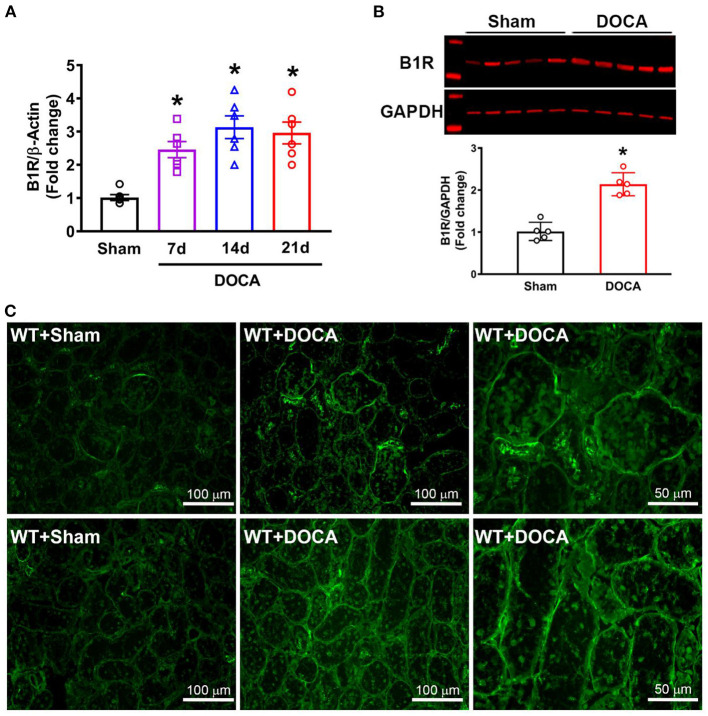
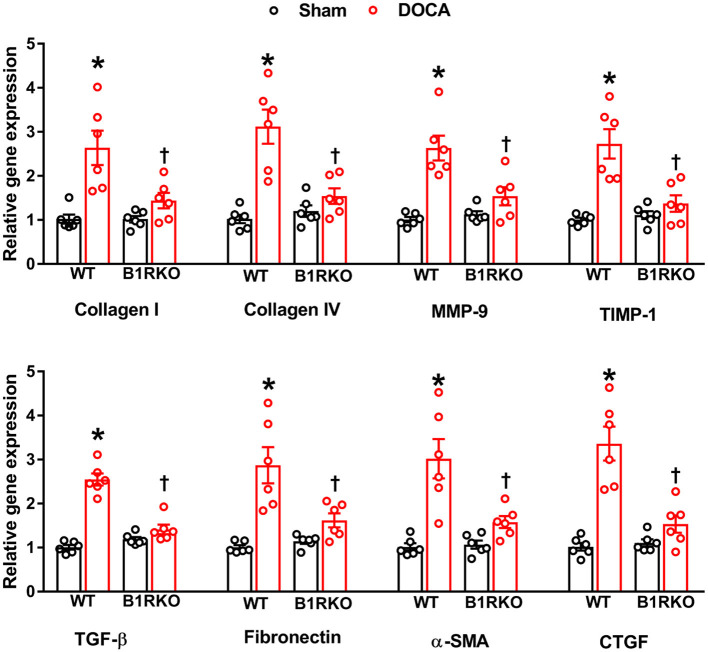
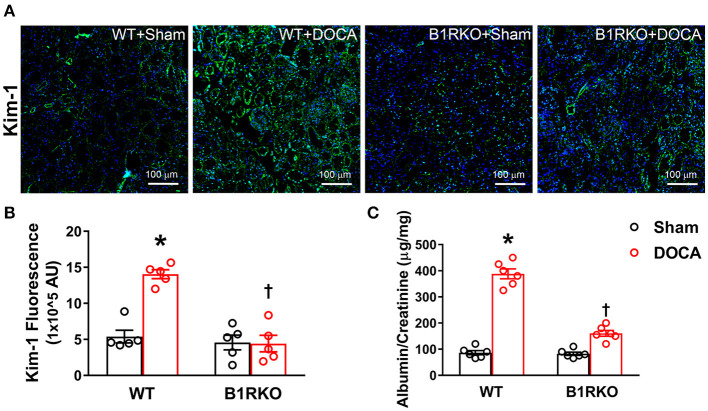
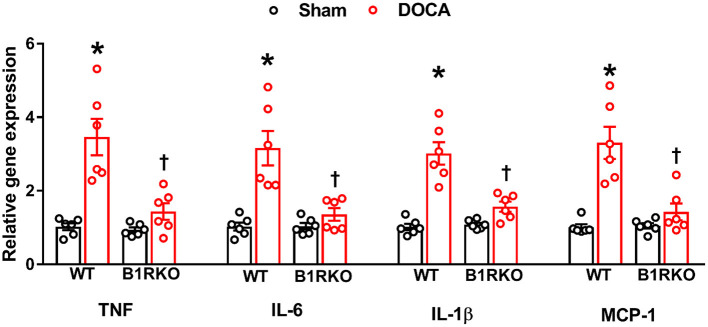
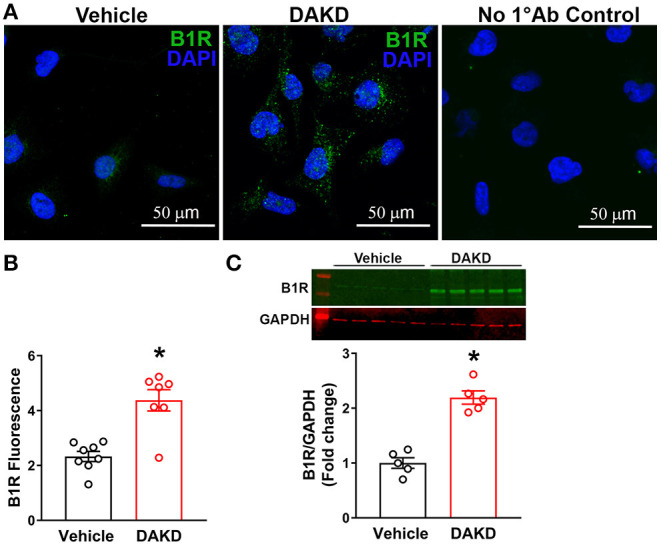
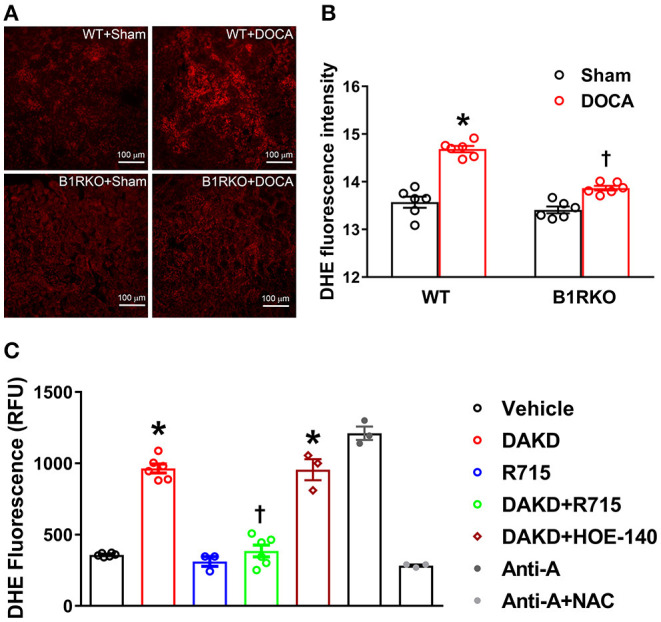
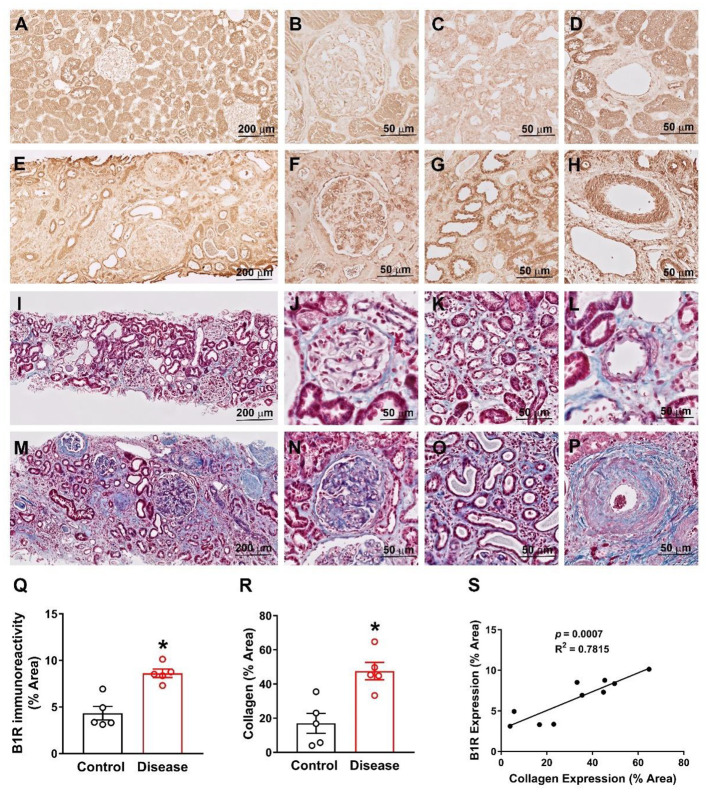
Despite many readily available therapies, hypertensive kidney disease remains the second most prevalent cause of end-stage renal disease after diabetes, and continues to burden patient populations and escalate morbidity and mortality rates. Kinin B1 receptor (B1R) activation has been shown to have a role in the development of hypertension, one of the major etiologies for chronic kidney disease. However, the role of B1R in hypertension induced renal injury and remodeling remains unexplored. Using a DOCA-salt-induced hypertensive mouse model, we investigated whether B1R deficiency reduces hypertensive renal injury and fibrosis. To further recognize the translational role of B1R, we examined the expression of B1R and its correlation with collagen deposition in renal biopsies from control and hypertensive kidney disease patients. Our data indicates that renal B1R expression was upregulated in the kidneys of DOCA-salt hypertensive mice. Genetic ablation of B1R protected the mice from DOCA-salt-induced renal injury and fibrosis by preventing inflammation and oxidative stress in the kidney. Cultured human proximal tubular epithelial cells expressed B1R and stimulation of B1R with an agonist resulted in increased oxidative stress. In human kidney biopsy samples, we found that the B1R immunoreactivity was not only significantly increased in hypertensive patients compared to normotensive patients, but also there is a positive correlation between B1R expression and renal fibrosis levels. Taken together, our results identify a critical role of B1R in the development of inflammation and fibrosis of the kidney in hypertension.
Keywords: hypertension; inflammation; kinin B1 receptor; oxidative stress; renal fibrosis.
Copyright © 2022 Basuli, Parekh, White, Thayyil and Sriramula.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Hypothalamic kinin B1 receptor mediates orexin system hyperactivity in neurogenic hypertension.Sci Rep. 2021 Oct 26;11(1):21050. doi: 10.1038/s41598-021-00522-0. Sci Rep. 2021. PMID: 34702886 Free PMC article.
-
Kinin B1 Receptor Mediates Bidirectional Interaction between Neuroinflammation and Oxidative Stress.Antioxidants (Basel). 2023 Jan 8;12(1):150. doi: 10.3390/antiox12010150. Antioxidants (Basel). 2023. PMID: 36671012 Free PMC article.
-
Renal protective role of bradykinin B1 receptor in stroke-prone spontaneously hypertensive rats.Hypertens Res. 2004 Jun;27(6):399-408. doi: 10.1291/hypres.27.399. Hypertens Res. 2004. PMID: 15253105
-
Kinin B1 Receptor Promotes Neurogenic Hypertension Through Activation of Centrally Mediated Mechanisms.Hypertension. 2017 Dec;70(6):1122-1131. doi: 10.1161/HYPERTENSIONAHA.117.09744. Epub 2017 Oct 16. Hypertension. 2017. PMID: 29038201 Free PMC article.
-
Kinin B1 Receptor Blockade Prevents Angiotensin II-induced Neuroinflammation and Oxidative Stress in Primary Hypothalamic Neurons.Cell Mol Neurobiol. 2020 Jul;40(5):845-857. doi: 10.1007/s10571-019-00778-1. Epub 2019 Dec 21. Cell Mol Neurobiol. 2020. PMID: 31865500 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources