

Precise gene models using long-read sequencing reveal a unique poly(A) signal in Giardia lamblia
- PMID: 35110372
- PMCID: PMC9014877
- DOI: 10.1261/rna.078793.121
Precise gene models using long-read sequencing reveal a unique poly(A) signal in Giardia lamblia
Abstract
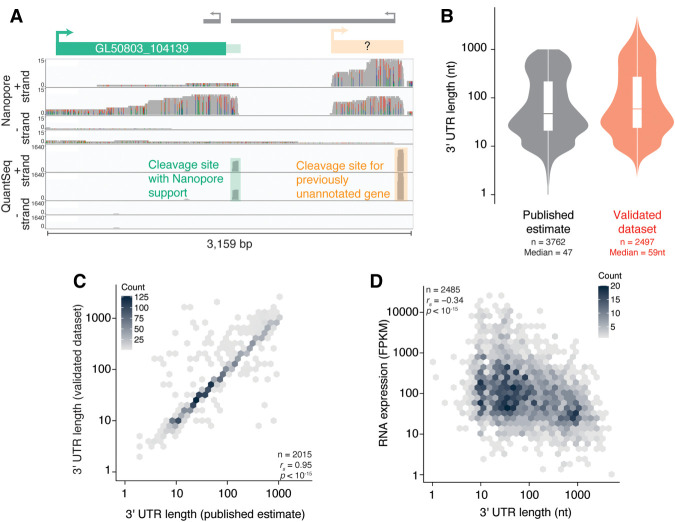
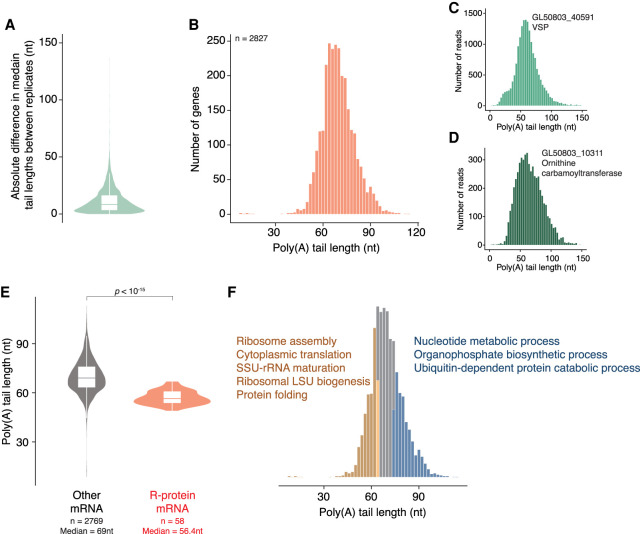
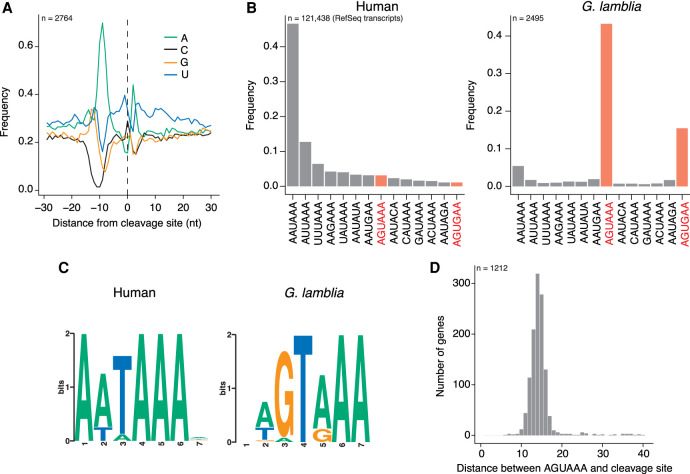
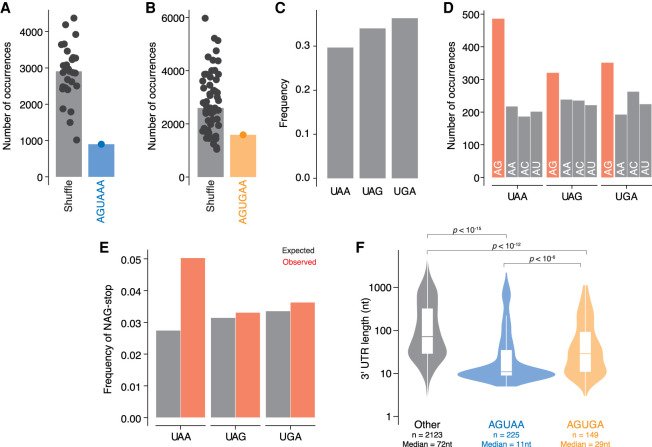
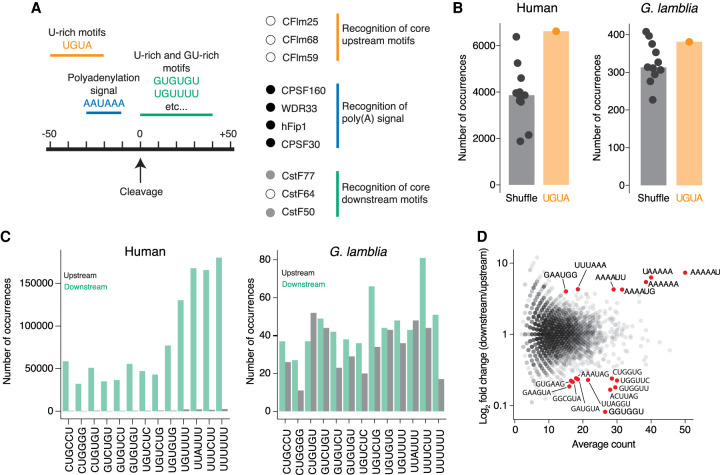
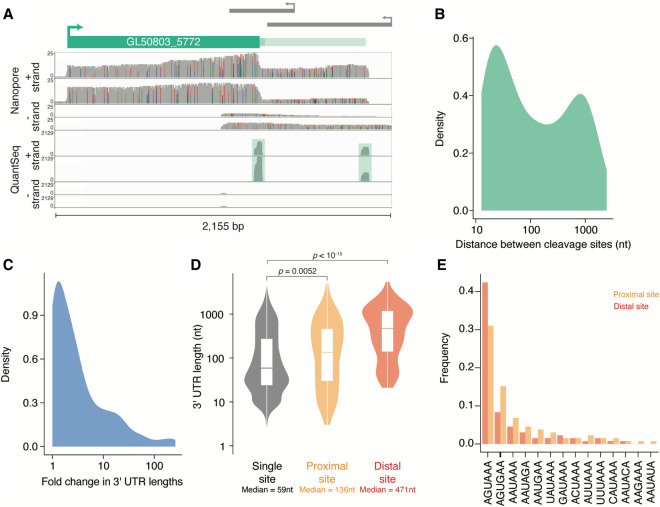
During pre-mRNA processing, the poly(A) signal is recognized by a protein complex that ensures precise cleavage and polyadenylation of the nascent transcript. The location of this cleavage event establishes the length and sequence of the 3' UTR of an mRNA, thus determining much of its post-transcriptional fate. Using long-read sequencing, we characterize the polyadenylation signal and related sequences surrounding Giardia lamblia cleavage sites for over 2600 genes. We find that G. lamblia uses an AGURAA poly(A) signal, which differs from the mammalian AAUAAA. We also describe how G. lamblia lacks common auxiliary elements found in other eukaryotes, along with the proteins that recognize them. Further, we identify 133 genes with evidence of alternative polyadenylation. These results suggest that despite pared-down cleavage and polyadenylation machinery, 3' end formation still appears to be an important regulatory step for gene expression in G. lamblia.
Keywords: 3′ UTR; Giardia lamblia; long-read sequencing; poly(A) site.
© 2022 Bilodeau et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures

Similar articles
-
Evolutionary dynamics of polyadenylation signals and their recognition strategies in protists.Genome Res. 2024 Oct 29;34(10):1570-1581. doi: 10.1101/gr.279526.124. Genome Res. 2024. PMID: 39327029
-
Implications of polyadenylation in health and disease.Nucleus. 2014;5(6):508-19. doi: 10.4161/nucl.36360. Epub 2014 Oct 31. Nucleus. 2014. PMID: 25484187 Free PMC article. Review.
-
mRNA Polyadenylation Machineries in Intestinal Protozoan Parasites.J Eukaryot Microbiol. 2020 May;67(3):306-320. doi: 10.1111/jeu.12781. Epub 2020 Jan 21. J Eukaryot Microbiol. 2020. PMID: 31898347
-
Developmentally regulated transcripts and evidence of differential mRNA processing in Giardia lamblia.Mol Biochem Parasitol. 1996 Oct 18;81(1):101-10. doi: 10.1016/0166-6851(96)02698-9. Mol Biochem Parasitol. 1996. PMID: 8892309
-
Structural biology of poly(A) site definition.Wiley Interdiscip Rev RNA. 2011 Sep-Oct;2(5):732-47. doi: 10.1002/wrna.88. Epub 2011 Apr 27. Wiley Interdiscip Rev RNA. 2011. PMID: 21823232 Free PMC article. Review.
Cited by
-
Spliceosomal introns in the diplomonad parasite Giardia duodenalis revisited.Microb Genom. 2023 Nov;9(11):001117. doi: 10.1099/mgen.0.001117. Microb Genom. 2023. PMID: 37934076 Free PMC article.
-
Evolutionary dynamics of polyadenylation signals and their recognition strategies in protists.Genome Res. 2024 Oct 29;34(10):1570-1581. doi: 10.1101/gr.279526.124. Genome Res. 2024. PMID: 39327029
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases