

p17-Modulated Hsp90/Cdc37 Complex Governs Oncolytic Avian Reovirus Replication by Chaperoning p17, Which Promotes Viral Protein Synthesis and Accumulation of Viral Proteins σC and σA in Viral Factories
- PMID: 35107368
- PMCID: PMC8941905
- DOI: 10.1128/jvi.00074-22
p17-Modulated Hsp90/Cdc37 Complex Governs Oncolytic Avian Reovirus Replication by Chaperoning p17, Which Promotes Viral Protein Synthesis and Accumulation of Viral Proteins σC and σA in Viral Factories
Erratum in
-
Correction for Huang et al., "p17-Modulated Hsp90/Cdc37 Complex Governs Oncolytic Avian Reovirus Replication by Chaperoning p17, Which Promotes Viral Protein Synthesis and Accumulation of Viral Proteins σC and σA in Viral Factories".J Virol. 2024 Jul 23;98(7):e0092024. doi: 10.1128/jvi.00920-24. Epub 2024 Jul 2. J Virol. 2024. PMID: 38953642 Free PMC article. No abstract available.
Abstract
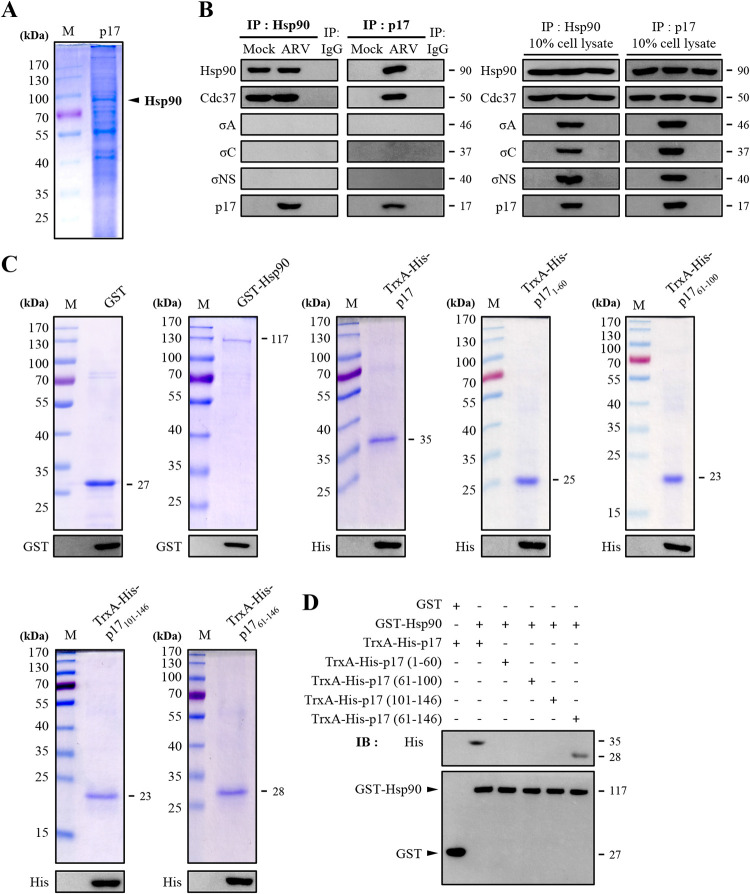
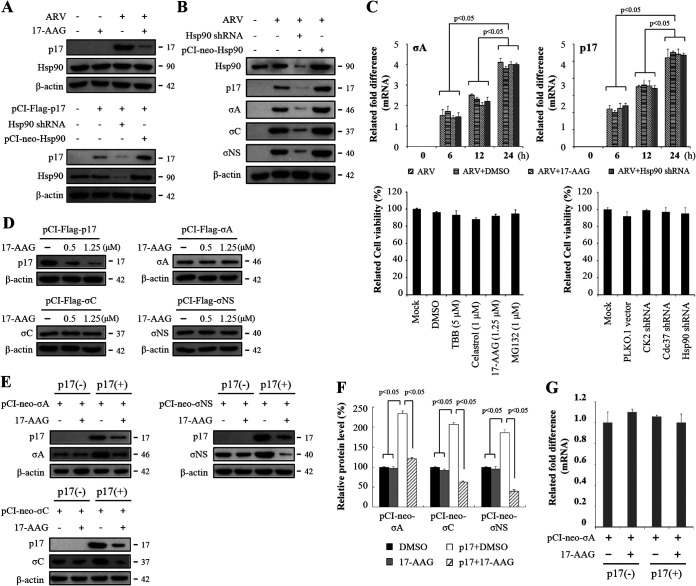
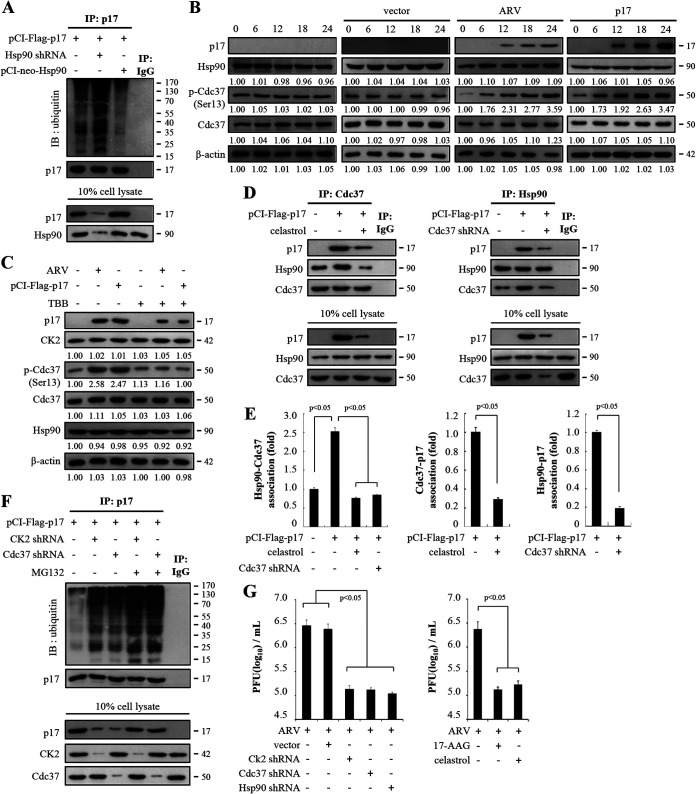
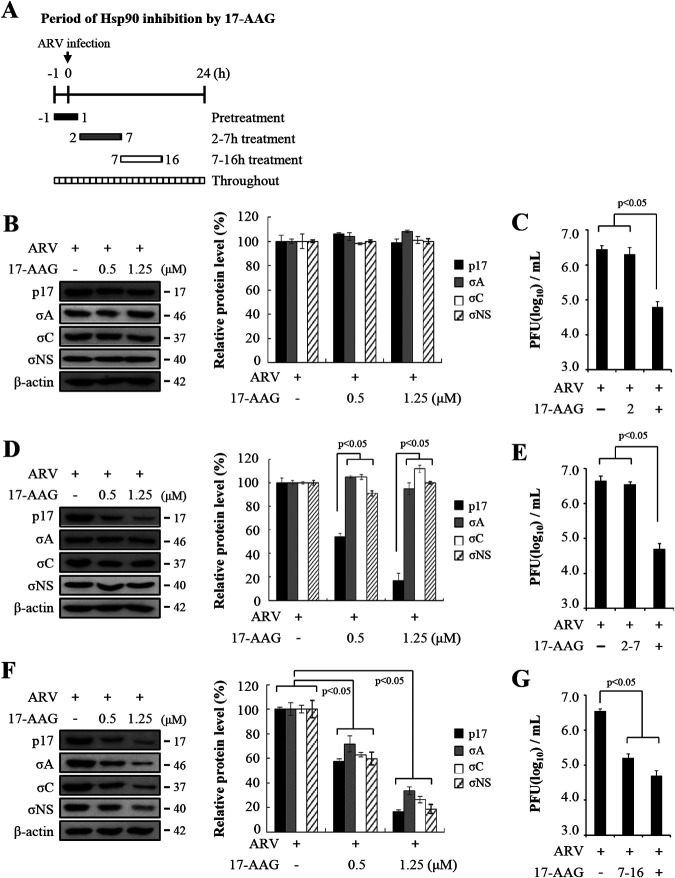
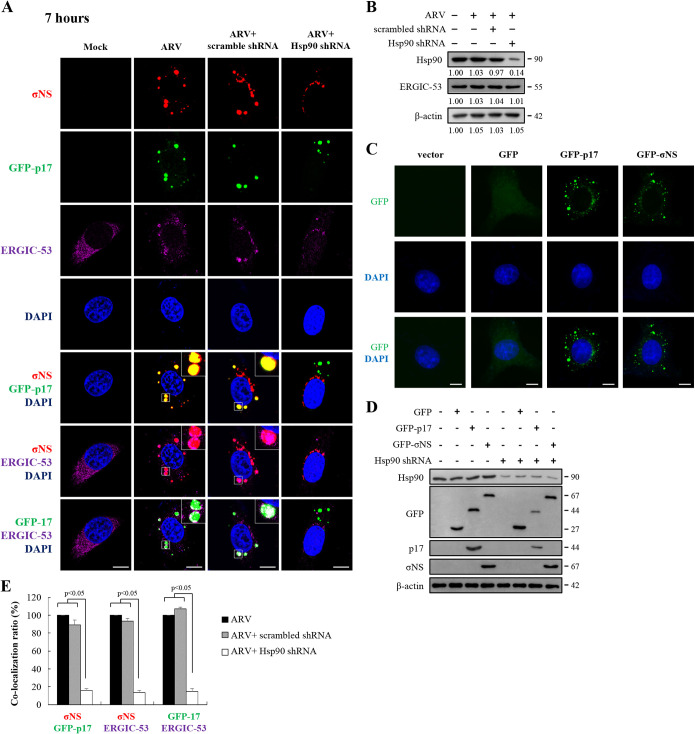
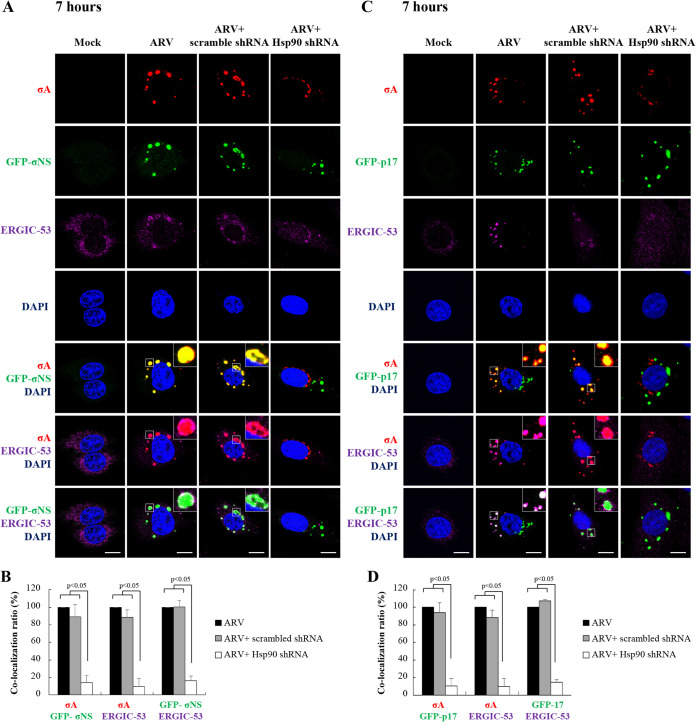
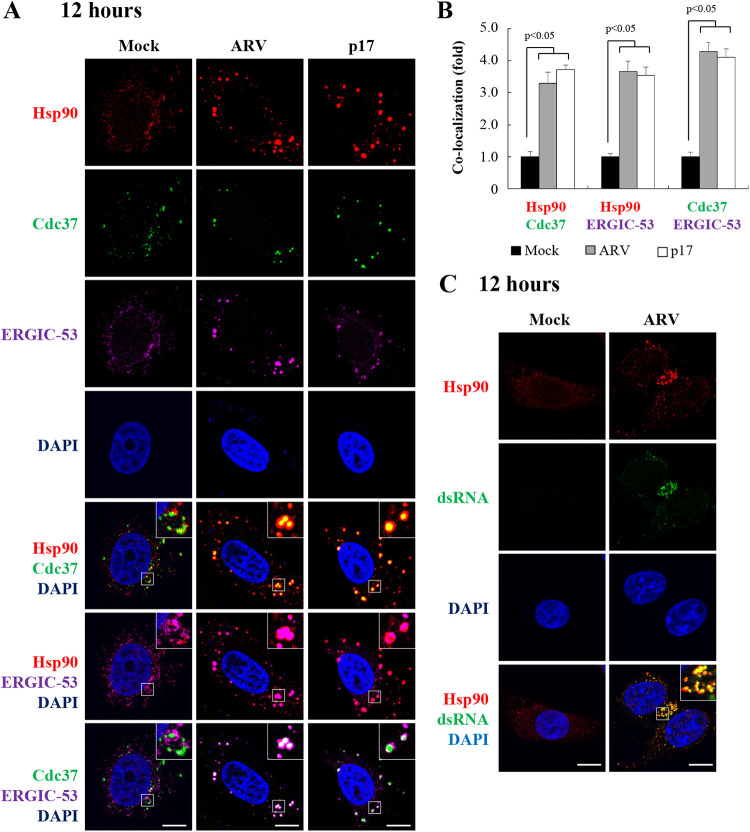
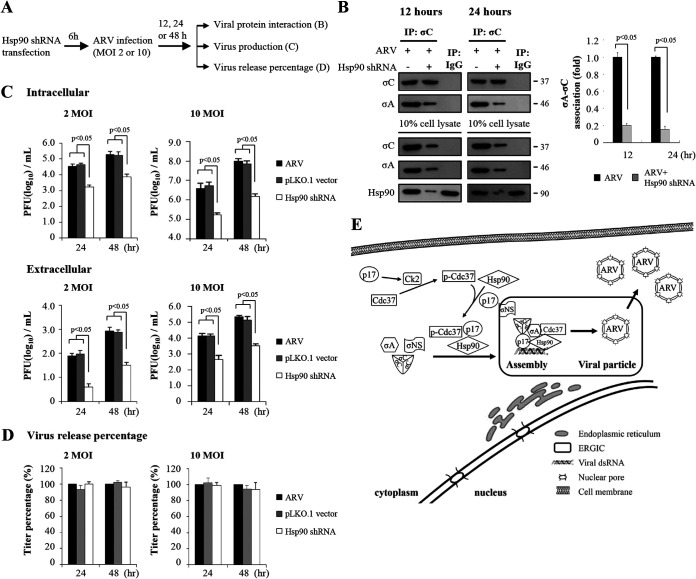
In this work we have determined that heat shock protein 90 (Hsp90) is essential for avian reovirus (ARV) replication by chaperoning the ARV p17 protein. p17 modulates the formation of the Hsp90/Cdc37 complex by phosphorylation of Cdc37, and this chaperone machinery protects p17 from ubiquitin-proteasome degradation. Inhibition of the Hsp90/Cdc37 complex by inhibitors (17-N-allylamino-17-demethoxygeldanamycin 17-AGG, and celastrol) or short hairpin RNAs (shRNAs) significantly reduced expression levels of viral proteins and virus yield, suggesting that the Hsp90/Cdc37 chaperone complex functions in virus replication. The expression levels of p17 were decreased at the examined time points (2 to 7 h and 7 to 16 h) in 17-AAG-treated cells in a dose-dependent manner while the expression levels of viral proteins σA, σC, and σNS were decreased at the examined time point (7 to 16 h). Interestingly, the expression levels of σC, σA, and σNS proteins increased along with coexpression of p17 protein. p17 together with the Hsp90/Cdc37 complex does not increase viral genome replication but enhances viral protein stability, maturation, and virus production. Virus factories of ARV are composed of nonstructural proteins σNS and μNS. We found that the Hsp90/Cdc37 chaperone complex plays an important role in accumulation of the outer-capsid protein σC, inner core protein σA, and nonstructural protein σNS of ARV in viral factories. Depletion of Hsp90 inhibited σA, σC, and p17 proteins colocalized with σNS in viral factories. This study provides novel insights into p17-modulated formation of the Hsp90/Cdc37 chaperone complex governing virus replication via stabilization and maturation of viral proteins and accumulation of viral proteins in viral factories for virus assembly. IMPORTANCE Molecular mechanisms that control stabilization of ARV proteins and the intermolecular interactions among inclusion components remain largely unknown. Here, we show that the ARV p17 is an Hsp90 client protein. The Hsp90/Cdc37 chaperone complex is essential for ARV replication by protecting p17 chaperone from ubiquitin-proteasome degradation. p17 modulates the formation of Hsp90/Cdc37 complex by phosphorylation of Cdc37, and this chaperone machinery protects p17 from ubiquitin-proteasome degradation, suggesting a feedback loop between p17 and the Hsp90/Cdc37 chaperone complex. p17 together with the Hsp90/Cdc37 complex does not increase viral genome replication but enhances viral protein stability and virus production. Depletion of Hsp90 prevented viral proteins σA, σC, and p17 from colocalizing with σNS in viral factories. Our findings elucidate that the Hsp90/Cdc37 complex chaperones p17, which, in turn, promotes the synthesis of viral proteins σA, σC, and σNS and facilitates accumulation of the outer-capsid protein σC and inner core protein σA in viral factories for virus assembly.
Keywords: Hsp90/Cdc37 chaperone machinery; avian reovirus; inner core protein σA; nonstructural protein p17; nonstructural protein σNS; outer-capsid protein σC; viral factory.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Molecular chaperone TRiC governs avian reovirus replication by protecting outer-capsid protein σC and inner core protein σA and non-structural protein σNS from ubiquitin- proteasome degradation.Vet Microbiol. 2022 Jan;264:109277. doi: 10.1016/j.vetmic.2021.109277. Epub 2021 Nov 10. Vet Microbiol. 2022. PMID: 34826648
-
Correction for Huang et al., "p17-Modulated Hsp90/Cdc37 Complex Governs Oncolytic Avian Reovirus Replication by Chaperoning p17, Which Promotes Viral Protein Synthesis and Accumulation of Viral Proteins σC and σA in Viral Factories".J Virol. 2024 Jul 23;98(7):e0092024. doi: 10.1128/jvi.00920-24. Epub 2024 Jul 2. J Virol. 2024. PMID: 38953642 Free PMC article. No abstract available.
-
The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/eIF2α signaling pathways.J Biol Chem. 2013 Feb 1;288(5):3571-84. doi: 10.1074/jbc.M112.390245. Epub 2012 Dec 11. J Biol Chem. 2013. PMID: 23233667 Free PMC article.
-
Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery.Cell Mol Life Sci. 2009 Jun;66(11-12):1840-9. doi: 10.1007/s00018-009-9152-0. Cell Mol Life Sci. 2009. PMID: 19387550 Free PMC article. Review.
-
Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery.J Hematol Oncol. 2018 Apr 27;11(1):59. doi: 10.1186/s13045-018-0602-8. J Hematol Oncol. 2018. PMID: 29699578 Free PMC article. Review.
Cited by
-
Oncolytic avian reovirus-sensitized tumor infiltrating CD8+ T cells triggering immunogenic apoptosis in gastric cancer.Cell Commun Signal. 2024 Oct 21;22(1):514. doi: 10.1186/s12964-024-01888-0. Cell Commun Signal. 2024. PMID: 39434159 Free PMC article.
-
The Oncolytic Avian Reovirus p17 Protein Inhibits Invadopodia Formation in Murine Melanoma Cancer Cells by Suppressing the FAK/Src Pathway and the Formation of theTKs5/NCK1 Complex.Viruses. 2024 Jul 17;16(7):1153. doi: 10.3390/v16071153. Viruses. 2024. PMID: 39066315 Free PMC article.
-
Oncolytic Avian Reovirus p17-Modulated Inhibition of mTORC1 by Enhancement of Endogenous mTORC1 Inhibitors Binding to mTORC1 To Disrupt Its Assembly and Accumulation on Lysosomes.J Virol. 2022 Sep 14;96(17):e0083622. doi: 10.1128/jvi.00836-22. Epub 2022 Aug 10. J Virol. 2022. PMID: 35946936 Free PMC article.
-
Oncolytic viruses-modulated immunogenic cell death, apoptosis and autophagy linking to virotherapy and cancer immune response.Front Cell Infect Microbiol. 2023 Mar 15;13:1142172. doi: 10.3389/fcimb.2023.1142172. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 37009515 Free PMC article. Review.
-
Identification and Functional Analyses of Host Proteins Interacting with the p17 Protein of Avian Reovirus.Viruses. 2022 Apr 25;14(5):892. doi: 10.3390/v14050892. Viruses. 2022. PMID: 35632635 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
