

Linkage disequilibrium between rare mutations
- PMID: 35100407
- PMCID: PMC8982034
- DOI: 10.1093/genetics/iyac004
Linkage disequilibrium between rare mutations
Abstract
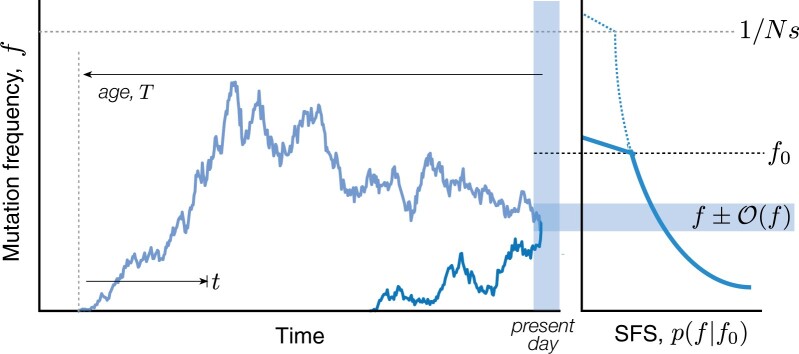
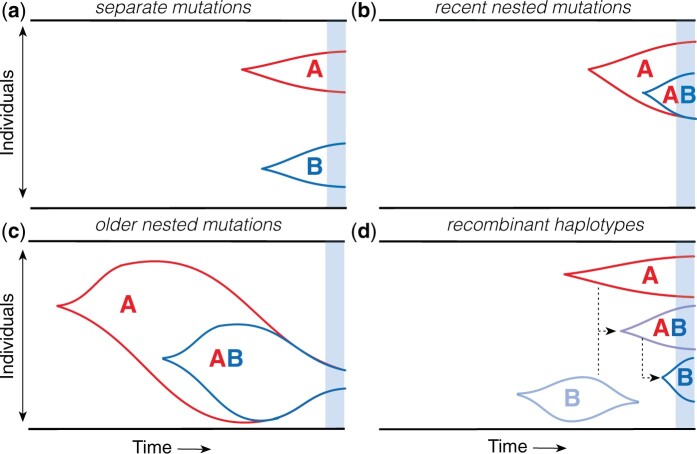
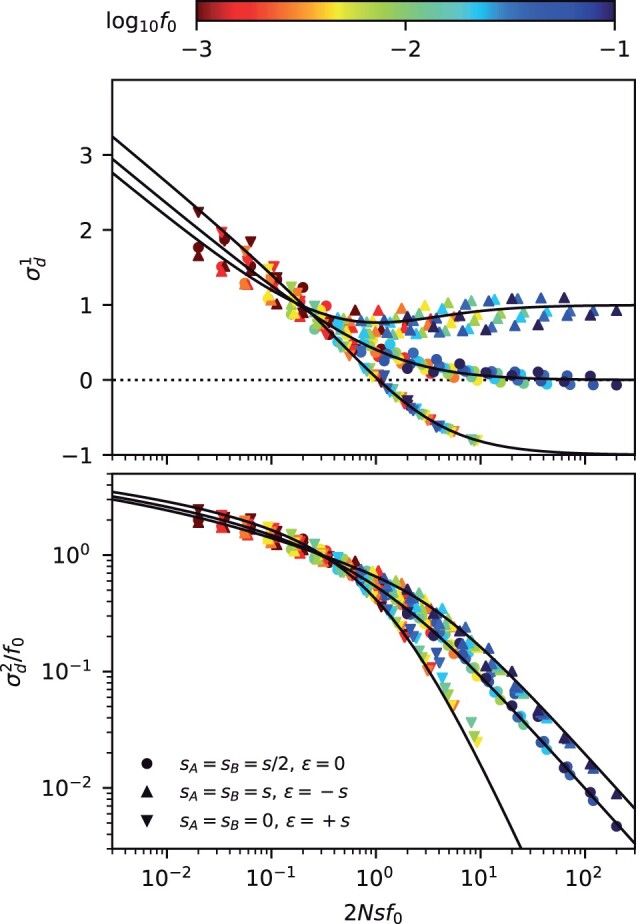
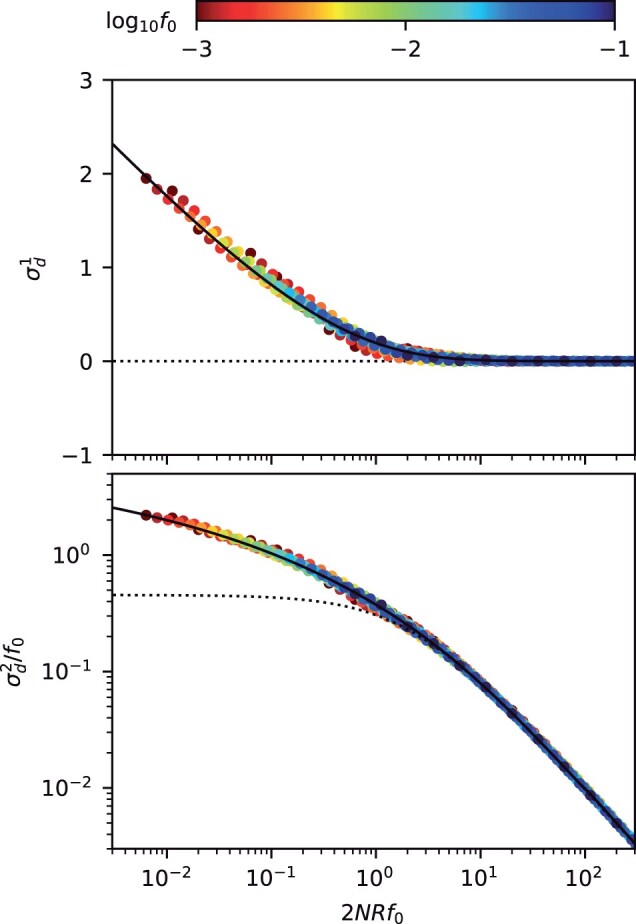
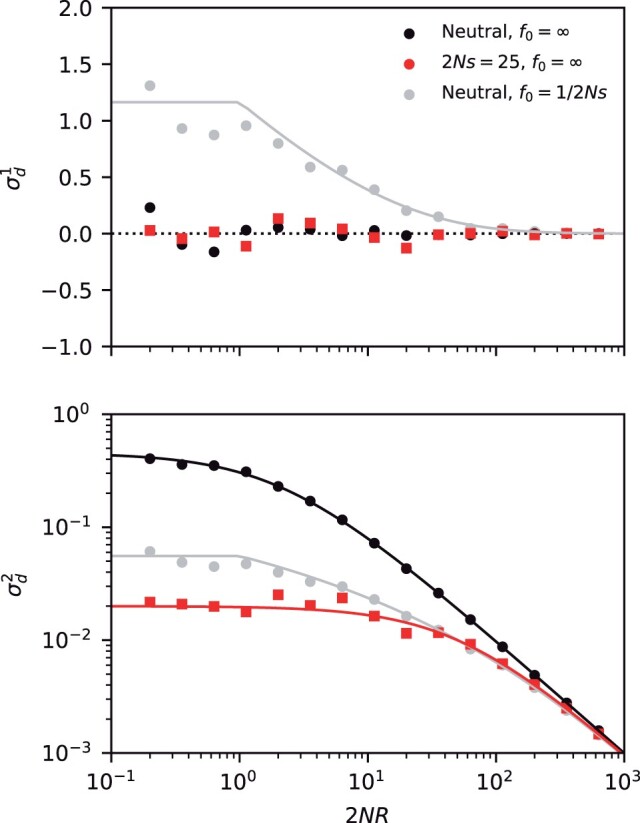
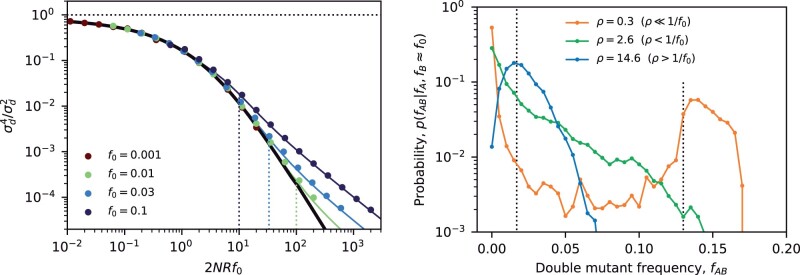
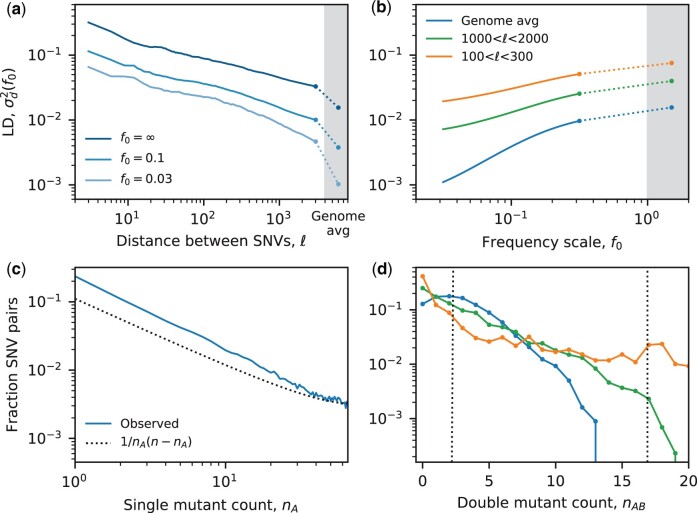
The statistical associations between mutations, collectively known as linkage disequilibrium, encode important information about the evolutionary forces acting within a population. Yet in contrast to single-site analogues like the site frequency spectrum, our theoretical understanding of linkage disequilibrium remains limited. In particular, little is currently known about how mutations with different ages and fitness costs contribute to expected patterns of linkage disequilibrium, even in simple settings where recombination and genetic drift are the major evolutionary forces. Here, I introduce a forward-time framework for predicting linkage disequilibrium between pairs of neutral and deleterious mutations as a function of their present-day frequencies. I show that the dynamics of linkage disequilibrium become much simpler in the limit that mutations are rare, where they admit a simple heuristic picture based on the trajectories of the underlying lineages. I use this approach to derive analytical expressions for a family of frequency-weighted linkage disequilibrium statistics as a function of the recombination rate, the frequency scale, and the additive and epistatic fitness costs of the mutations. I find that the frequency scale can have a dramatic impact on the shapes of the resulting linkage disequilibrium curves, reflecting the broad range of time scales over which these correlations arise. I also show that the differences between neutral and deleterious linkage disequilibrium are not purely driven by differences in their mutation frequencies and can instead display qualitative features that are reminiscent of epistasis. I conclude by discussing the implications of these results for recent linkage disequilibrium measurements in bacteria. This forward-time approach may provide a useful framework for predicting linkage disequilibrium across a range of evolutionary scenarios.
Keywords: epistasis; genetic drift; linkage disequilibrium; purifying selection; recombination.
© The Author(s) 2022. Published by Oxford University Press on behalf of Genetics Society of America. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Linkage equilibrium between rare mutations.Genetics. 2024 Nov 6;228(3):iyae145. doi: 10.1093/genetics/iyae145. Genetics. 2024. PMID: 39222343
-
Local fitness and epistatic effects lead to distinct patterns of linkage disequilibrium in protein-coding genes.Genetics. 2022 Jul 30;221(4):iyac097. doi: 10.1093/genetics/iyac097. Genetics. 2022. PMID: 35736370 Free PMC article.
-
Negative linkage disequilibrium between amino acid changing variants reveals interference among deleterious mutations in the human genome.PLoS Genet. 2021 Jul 28;17(7):e1009676. doi: 10.1371/journal.pgen.1009676. eCollection 2021 Jul. PLoS Genet. 2021. PMID: 34319975 Free PMC article.
-
The evolution of sex: empirical insights into the roles of epistasis and drift.Nat Rev Genet. 2007 Feb;8(2):139-49. doi: 10.1038/nrg1985. Nat Rev Genet. 2007. PMID: 17230200 Review.
-
Why sex and recombination?Cold Spring Harb Symp Quant Biol. 2009;74:187-95. doi: 10.1101/sqb.2009.74.030. Epub 2009 Nov 10. Cold Spring Harb Symp Quant Biol. 2009. PMID: 19903748 Review.
Cited by
-
A macroecological perspective on genetic diversity in the human gut microbiome.PLoS One. 2023 Jul 21;18(7):e0288926. doi: 10.1371/journal.pone.0288926. eCollection 2023. PLoS One. 2023. PMID: 37478102 Free PMC article.
-
Linkage equilibrium between rare mutations.bioRxiv [Preprint]. 2024 Apr 1:2024.03.28.587282. doi: 10.1101/2024.03.28.587282. bioRxiv. 2024. Update in: Genetics. 2024 Nov 6;228(3):iyae145. doi: 10.1093/genetics/iyae145 PMID: 38617331 Free PMC article. Updated. Preprint.
-
Dynamics of bacterial recombination in the human gut microbiome.PLoS Biol. 2024 Feb 8;22(2):e3002472. doi: 10.1371/journal.pbio.3002472. eCollection 2024 Feb. PLoS Biol. 2024. PMID: 38329938 Free PMC article.
-
Background Selection From Unlinked Sites Causes Nonindependent Evolution of Deleterious Mutations.Genome Biol Evol. 2024 Mar 2;16(3):evae050. doi: 10.1093/gbe/evae050. Genome Biol Evol. 2024. PMID: 38482769 Free PMC article.
-
Linkage equilibrium between rare mutations.Genetics. 2024 Nov 6;228(3):iyae145. doi: 10.1093/genetics/iyae145. Genetics. 2024. PMID: 39222343
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
