

Liver Fibrosis-From Mechanisms of Injury to Modulation of Disease
- PMID: 35087852
- PMCID: PMC8787129
- DOI: 10.3389/fmed.2021.814496
Liver Fibrosis-From Mechanisms of Injury to Modulation of Disease
Abstract
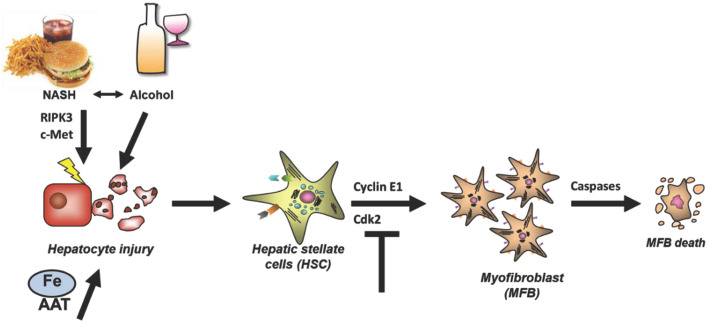
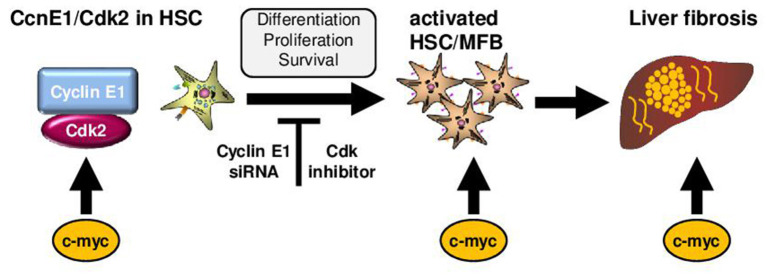
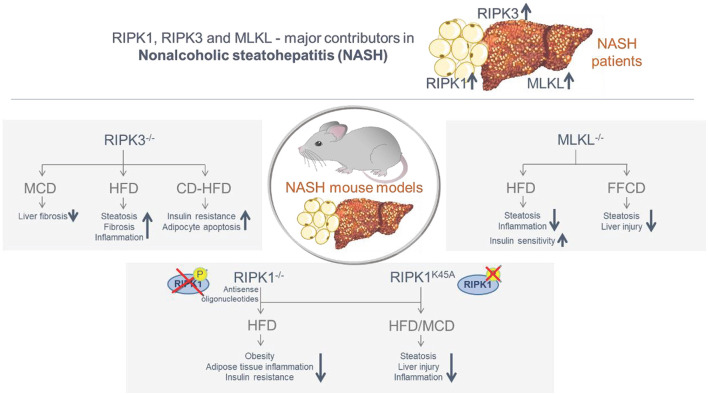
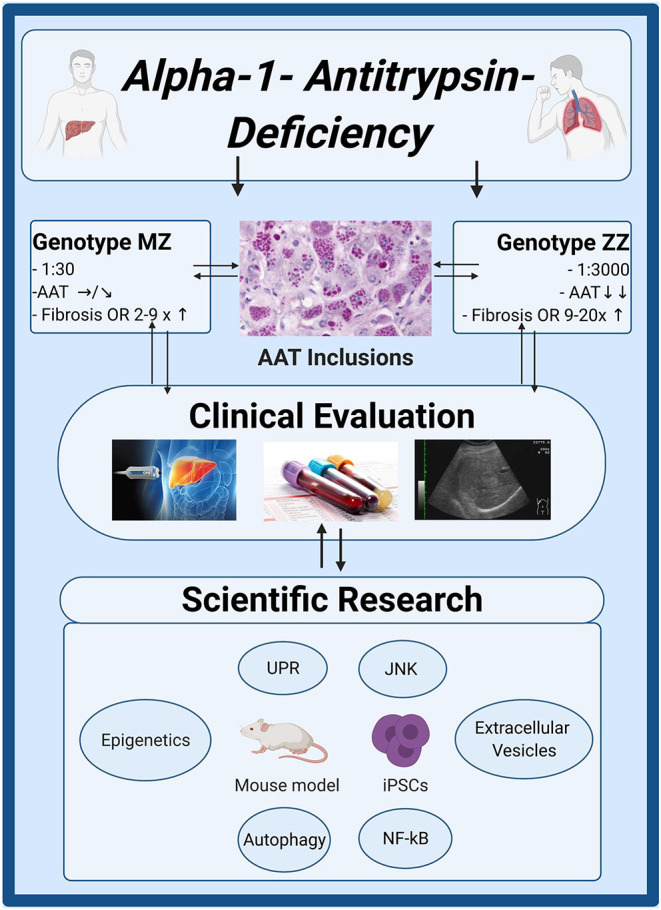
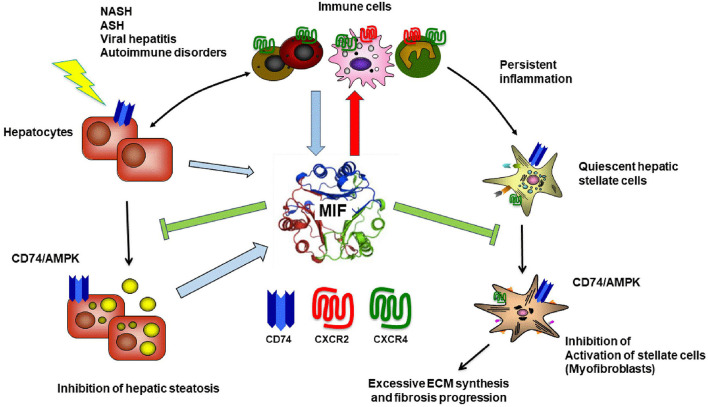
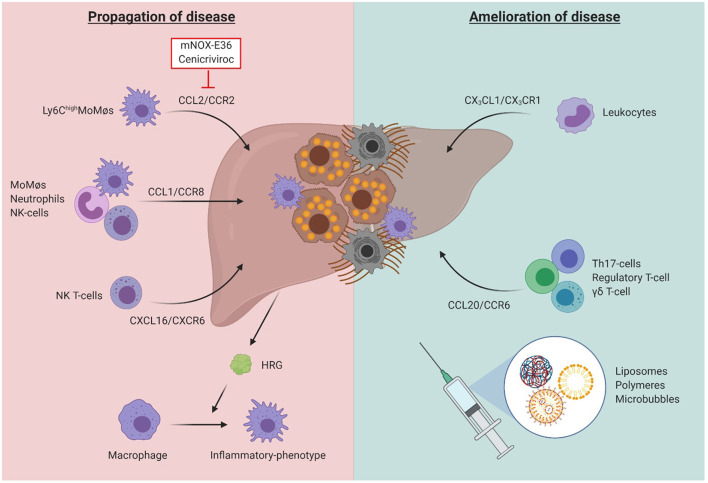
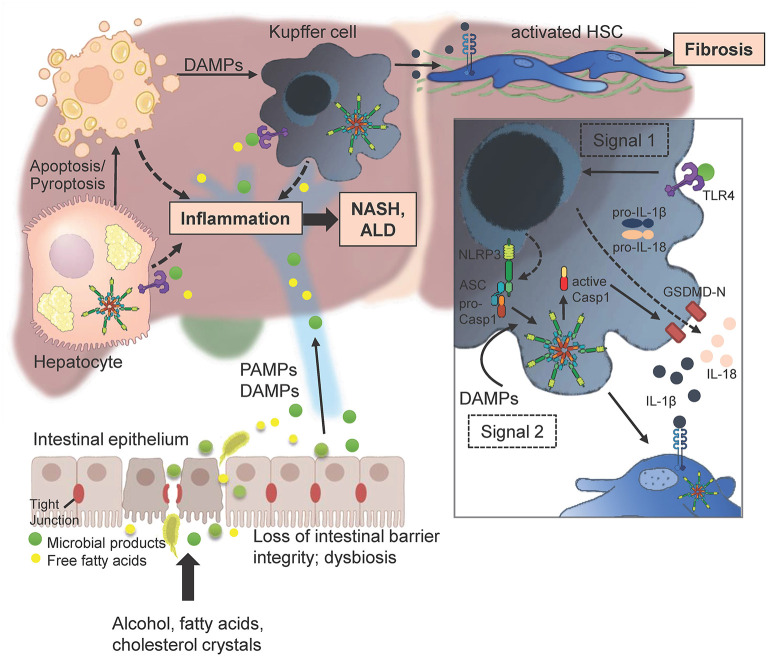
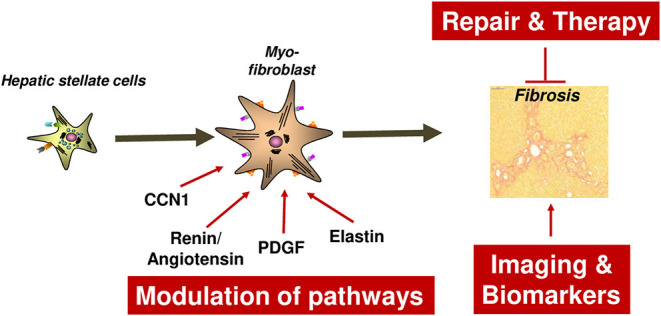
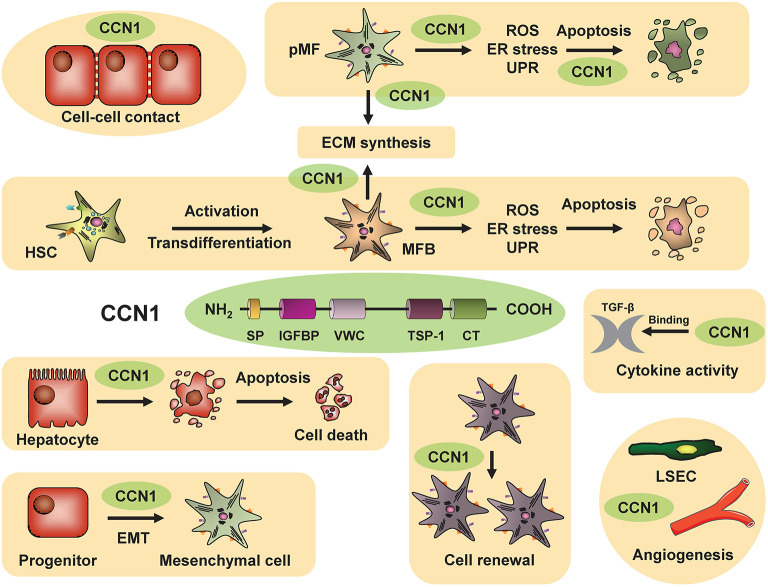
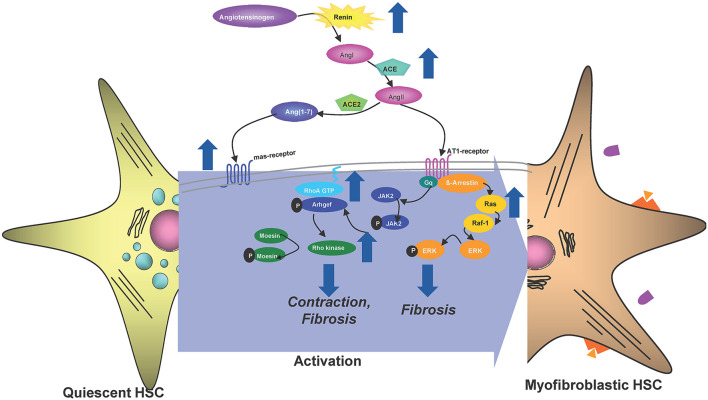
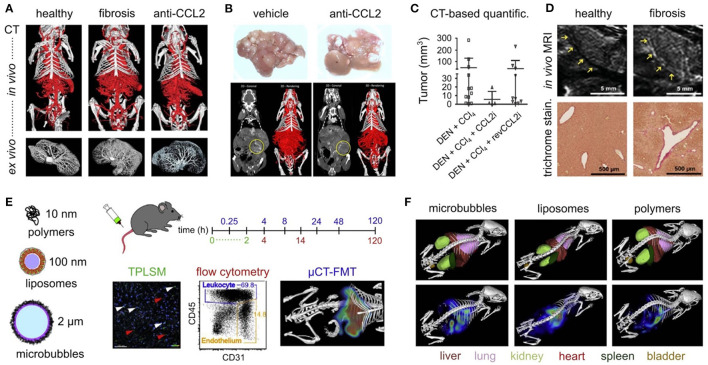
The Transregional Collaborative Research Center "Organ Fibrosis: From Mechanisms of Injury to Modulation of Disease" (referred to as SFB/TRR57) was funded for 13 years (2009-2021) by the German Research Council (DFG). This consortium was hosted by the Medical Schools of the RWTH Aachen University and Bonn University in Germany. The SFB/TRR57 implemented combined basic and clinical research to achieve detailed knowledge in three selected key questions: (i) What are the relevant mechanisms and signal pathways required for initiating organ fibrosis? (ii) Which immunological mechanisms and molecules contribute to organ fibrosis? and (iii) How can organ fibrosis be modulated, e.g., by interventional strategies including imaging and pharmacological approaches? In this review we will summarize the liver-related key findings of this consortium gained within the last 12 years on these three aspects of liver fibrogenesis. We will highlight the role of cell death and cell cycle pathways as well as nutritional and iron-related mechanisms for liver fibrosis initiation. Moreover, we will define and characterize the major immune cell compartments relevant for liver fibrogenesis, and finally point to potential signaling pathways and pharmacological targets that turned out to be suitable to develop novel approaches for improved therapy and diagnosis of liver fibrosis. In summary, this review will provide a comprehensive overview about the knowledge on liver fibrogenesis and its potential therapy gained by the SFB/TRR57 consortium within the last decade. The kidney-related research results obtained by the same consortium are highlighted in an article published back-to-back in Frontiers in Medicine.
Keywords: chemokines; cirrhosis; cytokines; extracellular matrix; hepatic stellate cell; hepatocytes; inflammation; resolution.
Copyright © 2022 Liedtke, Nevzorova, Luedde, Zimmermann, Kroy, Strnad, Berres, Bernhagen, Tacke, Nattermann, Spengler, Sauerbruch, Wree, Abdullah, Tolba, Trebicka, Lammers, Trautwein and Weiskirchen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Liver Fibrosis: From Basic Science towards Clinical Progress, Focusing on the Central Role of Hepatic Stellate Cells.Int J Mol Sci. 2024 Jul 18;25(14):7873. doi: 10.3390/ijms25147873. Int J Mol Sci. 2024. PMID: 39063116 Free PMC article. Review.
-
New Aspects of Kidney Fibrosis-From Mechanisms of Injury to Modulation of Disease.Front Med (Lausanne). 2022 Jan 12;8:814497. doi: 10.3389/fmed.2021.814497. eCollection 2021. Front Med (Lausanne). 2022. PMID: 35096904 Free PMC article. Review.
-
The carbon tetrachloride model in mice.Lab Anim. 2015 Apr;49(1 Suppl):4-11. doi: 10.1177/0023677215571192. Lab Anim. 2015. PMID: 25835733
-
Mechanisms of cardiovascular complications in chronic kidney disease: research focus of the Transregional Research Consortium SFB TRR219 of the University Hospital Aachen (RWTH) and the Saarland University.Clin Res Cardiol. 2018 Aug;107(Suppl 2):120-126. doi: 10.1007/s00392-018-1260-0. Epub 2018 May 4. Clin Res Cardiol. 2018. PMID: 29728829 Review.
-
Liver fibrosis: from the bench to clinical targets.Dig Liver Dis. 2004 Apr;36(4):231-42. doi: 10.1016/j.dld.2004.01.003. Dig Liver Dis. 2004. PMID: 15115333 Review.
Cited by
-
Liver Fibrosis: From Basic Science towards Clinical Progress, Focusing on the Central Role of Hepatic Stellate Cells.Int J Mol Sci. 2024 Jul 18;25(14):7873. doi: 10.3390/ijms25147873. Int J Mol Sci. 2024. PMID: 39063116 Free PMC article. Review.
-
A model of hepatic steatosis with declined viability and function in a liver-organ-on-a-chip.Sci Rep. 2023 Oct 9;13(1):17019. doi: 10.1038/s41598-023-44198-0. Sci Rep. 2023. PMID: 37813918 Free PMC article.
-
Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications.Cells. 2023 Jun 24;12(13):1712. doi: 10.3390/cells12131712. Cells. 2023. PMID: 37443746 Free PMC article. Review.
-
Sjögren's Syndrome-Related Organs Fibrosis: Hypotheses and Realities.J Clin Med. 2022 Jun 20;11(12):3551. doi: 10.3390/jcm11123551. J Clin Med. 2022. PMID: 35743618 Free PMC article. Review.
-
Wharton's jelly mesenchymal stem cells: a concise review of their secretome and prospective clinical applications.Front Cell Dev Biol. 2023 Jun 27;11:1211217. doi: 10.3389/fcell.2023.1211217. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37440921 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources