

Regulation of TNF-Induced Osteoclast Differentiation
- PMID: 35011694
- PMCID: PMC8750957
- DOI: 10.3390/cells11010132
Regulation of TNF-Induced Osteoclast Differentiation
Abstract
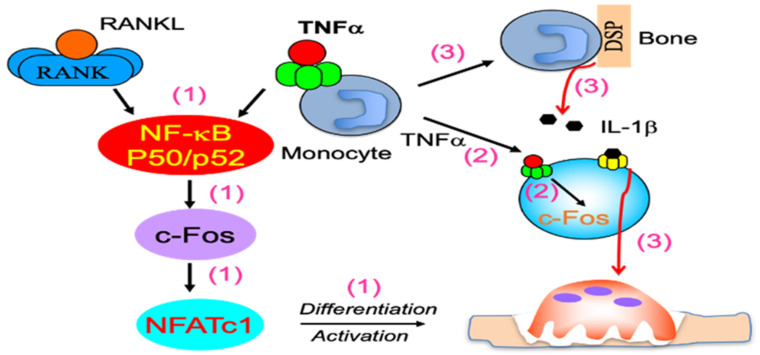
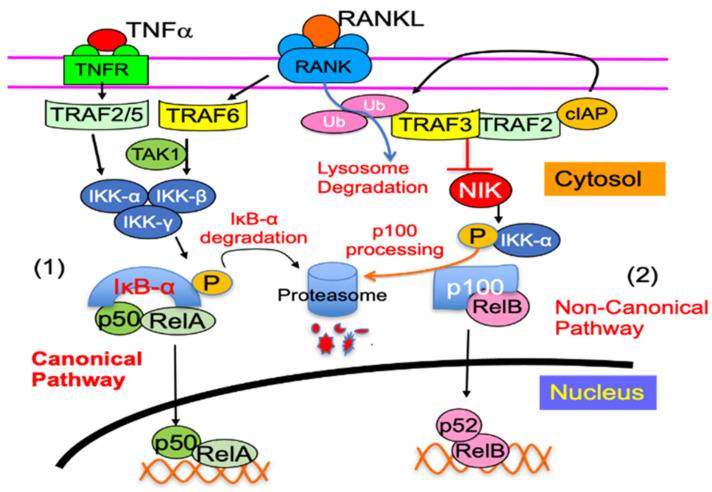
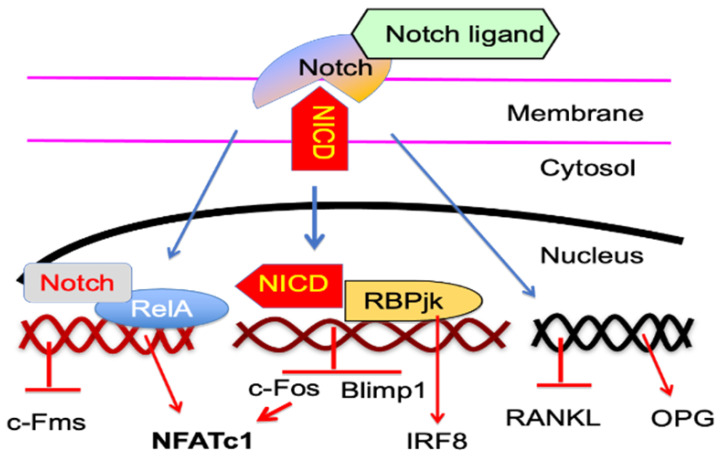
Increased osteoclast (OC) differentiation and activity is the critical event that results in bone loss and joint destruction in common pathological bone conditions, such as osteoporosis and rheumatoid arthritis (RA). RANKL and its decoy receptor, osteoprotegerin (OPG), control OC differentiation and activity. However, there is a specific concern of a rebound effect of denosumab discontinuation in treating osteoporosis. TNFα can induce OC differentiation that is independent of the RANKL/RANK system. In this review, we discuss the factors that negatively and positively regulate TNFα induction of OC formation, and the mechanisms involved to inform the design of new anti-resorptive agents for the treatment of bone conditions with enhanced OC formation. Similar to, and being independent of, RANKL, TNFα recruits TNF receptor-associated factors (TRAFs) to sequentially activate transcriptional factors NF-κB p50 and p52, followed by c-Fos, and then NFATc1 to induce OC differentiation. However, induction of OC formation by TNFα alone is very limited, since it also induces many inhibitory proteins, such as TRAF3, p100, IRF8, and RBP-j. TNFα induction of OC differentiation is, however, versatile, and Interleukin-1 or TGFβ1 can enhance TNFα-induced OC formation through a mechanism which is independent of RANKL, TRAF6, and/or NF-κB. However, TNFα polarized macrophages also produce anabolic factors, including insulin such as 6 peptide and Jagged1, to slow down bone loss in the pathological conditions. Thus, the development of novel approaches targeting TNFα signaling should focus on its downstream molecules that do not affect its anabolic effect.
Keywords: TNF receptor-associated factor 3 (TRAF3); interferon-regulatory factor 8 (IRF8); interleukin-1β (IL-1β); nuclear factor-kappa B (NF-κB); osteoclast; osteoprotegerin (OPG); receptor activator of NF-κB ligand (RANKL); recombination signal–binding protein jκ (RBPjκ); transforming growth factor-β1 (TGF-β1); tumor necrosis factor alpha (TNFα).
Conflict of interest statement
The authors declare no conflict of interest. The content is solely the responsibility of the authors, and does not necessarily represent the official views of the funding source.
Figures
Similar articles
-
NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1.J Biol Chem. 2007 Jun 22;282(25):18245-18253. doi: 10.1074/jbc.M610701200. Epub 2007 May 7. J Biol Chem. 2007. PMID: 17485464
-
TNF Induction of NF-κB RelB Enhances RANKL-Induced Osteoclastogenesis by Promoting Inflammatory Macrophage Differentiation but also Limits It through Suppression of NFATc1 Expression.PLoS One. 2015 Aug 19;10(8):e0135728. doi: 10.1371/journal.pone.0135728. eCollection 2015. PLoS One. 2015. PMID: 26287732 Free PMC article.
-
RANKL cytokine enhances TNF-induced osteoclastogenesis independently of TNF receptor associated factor (TRAF) 6 by degrading TRAF3 in osteoclast precursors.J Biol Chem. 2017 Jun 16;292(24):10169-10179. doi: 10.1074/jbc.M116.771816. Epub 2017 Apr 24. J Biol Chem. 2017. PMID: 28438834 Free PMC article.
-
Involvement of receptor activator of NFkappaB ligand and tumor necrosis factor-alpha in bone destruction in rheumatoid arthritis.Bone. 2002 Feb;30(2):340-6. doi: 10.1016/s8756-3282(01)00682-2. Bone. 2002. PMID: 11856640 Review.
-
Osteoclast differentiation by RANKL and OPG signaling pathways.J Bone Miner Metab. 2021 Jan;39(1):19-26. doi: 10.1007/s00774-020-01162-6. Epub 2020 Oct 20. J Bone Miner Metab. 2021. PMID: 33079279 Review.
Cited by
-
Trehalose Rescues Postmenopausal Osteoporosis Induced by Ovariectomy through Alleviating Osteoblast Pyroptosis via Promoting Autophagy.Biomedicines. 2024 Sep 29;12(10):2224. doi: 10.3390/biomedicines12102224. Biomedicines. 2024. PMID: 39457537 Free PMC article.
-
Modulation of fracture healing by senescence-associated secretory phenotype (SASP): a narrative review of the current literature.Eur J Med Res. 2024 Jan 9;29(1):38. doi: 10.1186/s40001-023-01604-7. Eur J Med Res. 2024. PMID: 38195489 Free PMC article. Review.
-
[Role and mechanism of macrophage-mediated osteoimmune in osteonecrosis of the femoral head].Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2024 Jan 15;38(1):119-124. doi: 10.7507/1002-1892.202308026. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2024. PMID: 38225851 Free PMC article. Chinese.
-
Babam2 negatively regulates osteoclastogenesis by interacting with Hey1 to inhibit Nfatc1 transcription.Int J Biol Sci. 2022 Jul 11;18(11):4482-4496. doi: 10.7150/ijbs.72487. eCollection 2022. Int J Biol Sci. 2022. PMID: 35864959 Free PMC article.
-
Betulinic Acid Attenuates Osteoarthritis via Limiting NLRP3 Inflammasome Activation to Decrease Interleukin-1β Maturation and Secretion.Mediators Inflamm. 2023 Sep 25;2023:3706421. doi: 10.1155/2023/3706421. eCollection 2023. Mediators Inflamm. 2023. PMID: 37789884 Free PMC article.
References
-
- Eriksen E.F., Hodgson S.F., Eastell R., Riggs B.L., Cedel S.L., O’Fallon W.M. Cancellous bone remodeling in type i (postmenopausal) osteoporosis: Quantitative assessment of rates of formation, resorption, and bone loss at tissue and cellular levels. J. Bone Miner. Res. 1990;5:311–319. doi: 10.1002/jbmr.5650050402. - DOI - PubMed
-
- Redlich K., Hayer S., Maier A., Dunstan C.R., Tohidast-Akrad M., Lang S., Türk B., Pietschmann P., Woloszczuk W., Haralambous S., et al. Tumor necrosis factor α-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum. 2002;46:785–792. doi: 10.1002/art.10097. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
