

Lipid bilayer induces contraction of the denatured state ensemble of a helical-bundle membrane protein
- PMID: 34969836
- PMCID: PMC8740594
- DOI: 10.1073/pnas.2109169119
Lipid bilayer induces contraction of the denatured state ensemble of a helical-bundle membrane protein
Erratum in
-
Correction to Gaffney et al., Lipid bilayer induces contraction of the denatured state ensemble of a helical-bundle membrane protein.Proc Natl Acad Sci U S A. 2024 Sep 17;121(38):e2415554121. doi: 10.1073/pnas.2415554121. Epub 2024 Sep 10. Proc Natl Acad Sci U S A. 2024. PMID: 39255002 Free PMC article. No abstract available.
Abstract
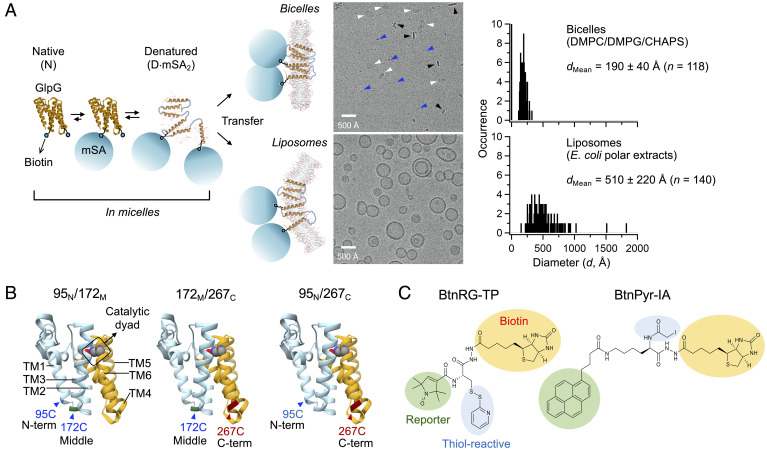
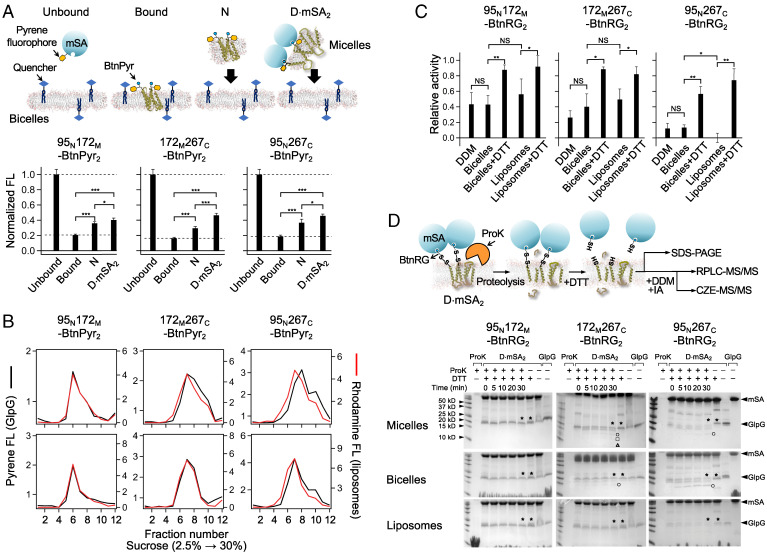
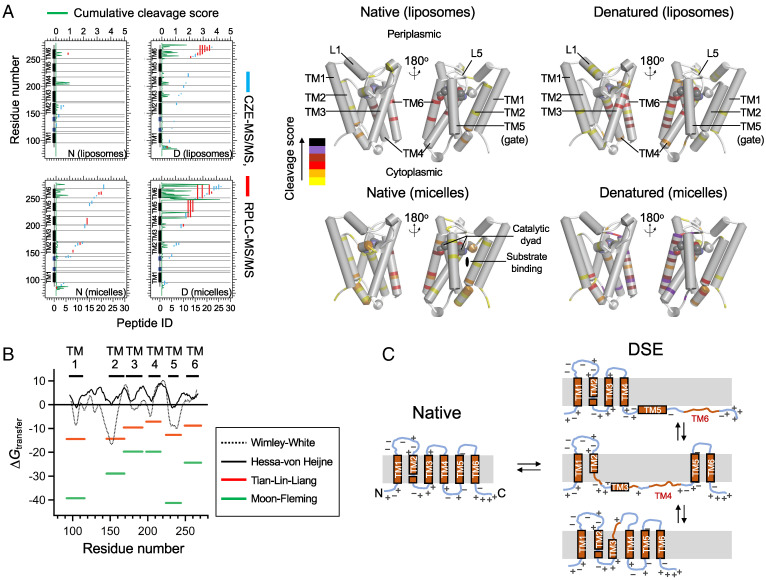
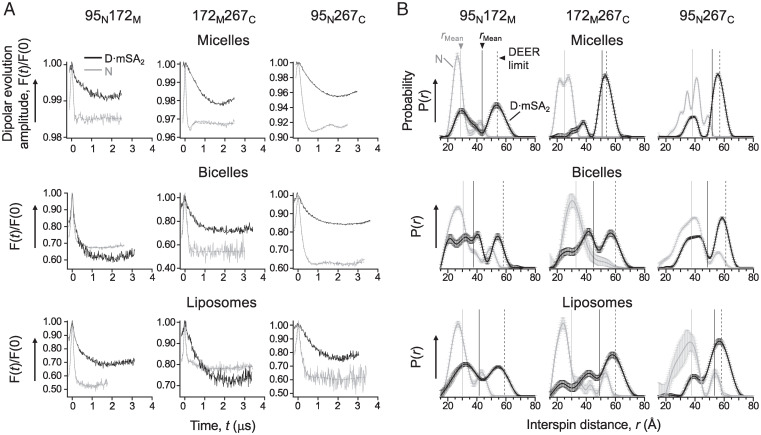
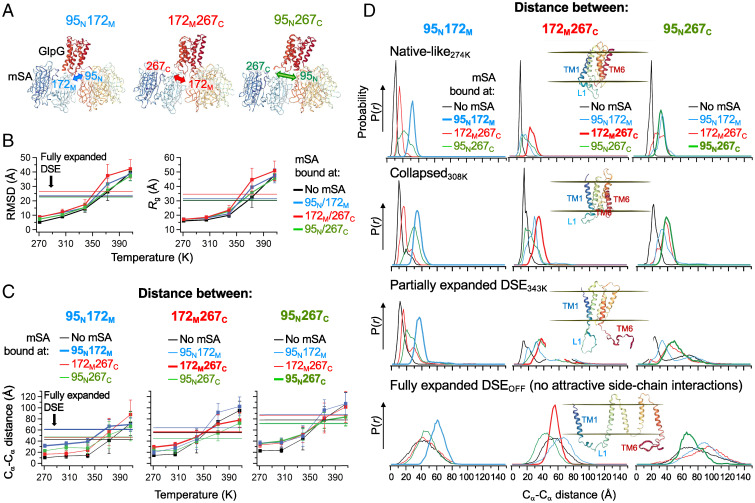
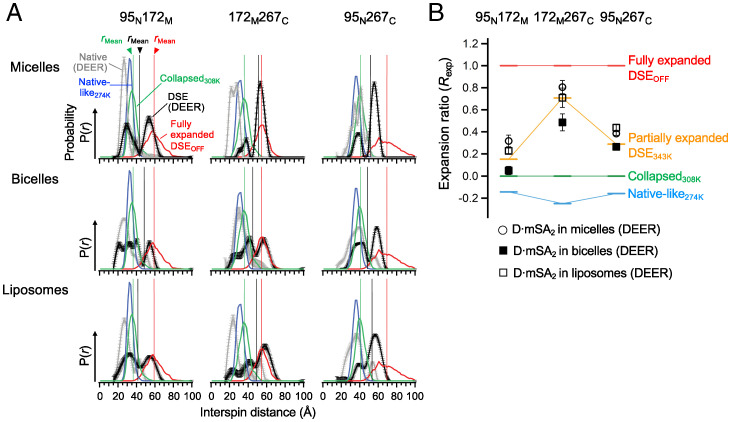
Defining the denatured state ensemble (DSE) and disordered proteins is essential to understanding folding, chaperone action, degradation, and translocation. As compared with water-soluble proteins, the DSE of membrane proteins is much less characterized. Here, we measure the DSE of the helical membrane protein GlpG of Escherichia coli (E. coli) in native-like lipid bilayers. The DSE was obtained using our steric trapping method, which couples denaturation of doubly biotinylated GlpG to binding of two streptavidin molecules. The helices and loops are probed using limited proteolysis and mass spectrometry, while the dimensions are determined using our paramagnetic biotin derivative and double electron-electron resonance spectroscopy. These data, along with our Upside simulations, identify the DSE as being highly dynamic, involving the topology changes and unfolding of some of the transmembrane (TM) helices. The DSE is expanded relative to the native state but only to 15 to 75% of the fully expanded condition. The degree of expansion depends on the local protein packing and the lipid composition. E. coli's lipid bilayer promotes the association of TM helices in the DSE and, probably in general, facilitates interhelical interactions. This tendency may be the outcome of a general lipophobic effect of proteins within the cell membranes.
Keywords: GlpG; Upside simulation; denatured state; membrane protein folding; steric trapping.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
Steric trapping strategy for studying the folding of helical membrane proteins.Methods. 2024 May;225:1-12. doi: 10.1016/j.ymeth.2024.02.007. Epub 2024 Feb 29. Methods. 2024. PMID: 38428472 Review.
-
Steric trapping reveals a cooperativity network in the intramembrane protease GlpG.Nat Chem Biol. 2016 May;12(5):353-360. doi: 10.1038/nchembio.2048. Epub 2016 Mar 21. Nat Chem Biol. 2016. PMID: 26999782 Free PMC article.
-
Topological constraints and modular structure in the folding and functional motions of GlpG, an intramembrane protease.Proc Natl Acad Sci U S A. 2016 Feb 23;113(8):2098-103. doi: 10.1073/pnas.1524027113. Epub 2016 Feb 8. Proc Natl Acad Sci U S A. 2016. PMID: 26858402 Free PMC article.
-
Measuring transmembrane helix interaction strengths in lipid bilayers using steric trapping.Methods Mol Biol. 2013;1063:37-56. doi: 10.1007/978-1-62703-583-5_3. Methods Mol Biol. 2013. PMID: 23975771 Free PMC article.
-
Lipids modulate the insertion and folding of the nascent chains of alpha helical membrane proteins.Biochem Soc Trans. 2018 Oct 19;46(5):1355-1366. doi: 10.1042/BST20170424. Epub 2018 Sep 6. Biochem Soc Trans. 2018. PMID: 30190329 Review.
Cited by
-
Factors That Control the Force Needed to Unfold a Membrane Protein in Silico Depend on the Mode of Denaturation.Int J Mol Sci. 2023 Jan 31;24(3):2654. doi: 10.3390/ijms24032654. Int J Mol Sci. 2023. PMID: 36768981 Free PMC article.
-
Methods to study folding of alpha-helical membrane proteins in lipids.Open Biol. 2022 Jul;12(7):220054. doi: 10.1098/rsob.220054. Epub 2022 Jul 20. Open Biol. 2022. PMID: 35855589 Free PMC article. Review.
-
Mechanistic Insight into the Mechanical Unfolding of the Integral Membrane Diacylglycerol Kinase.JACS Au. 2024 Mar 16;4(4):1422-1435. doi: 10.1021/jacsau.3c00829. eCollection 2024 Apr 22. JACS Au. 2024. PMID: 38665647 Free PMC article.
-
Steric trapping strategy for studying the folding of helical membrane proteins.Methods. 2024 May;225:1-12. doi: 10.1016/j.ymeth.2024.02.007. Epub 2024 Feb 29. Methods. 2024. PMID: 38428472 Review.
-
Folding speeds of helical membrane proteins.Biochem Soc Trans. 2024 Feb 28;52(1):491-501. doi: 10.1042/BST20231315. Biochem Soc Trans. 2024. PMID: 38385525 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
