

Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress
- PMID: 34943836
- PMCID: PMC8699655
- DOI: 10.3390/cells10123328
Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress
Abstract
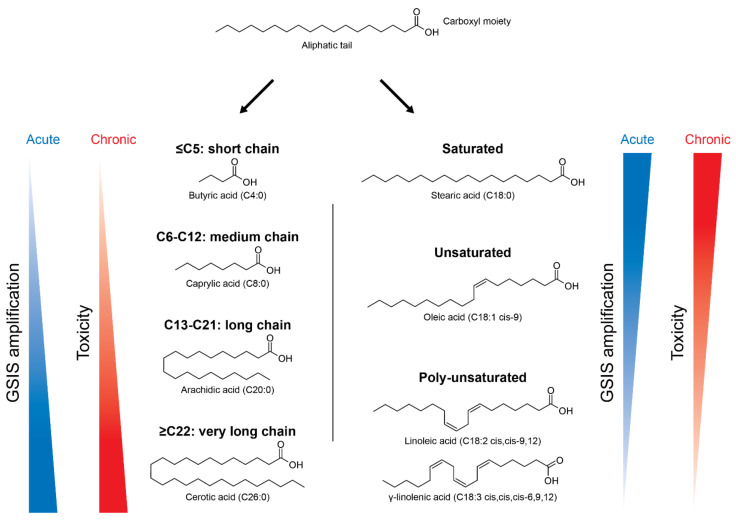
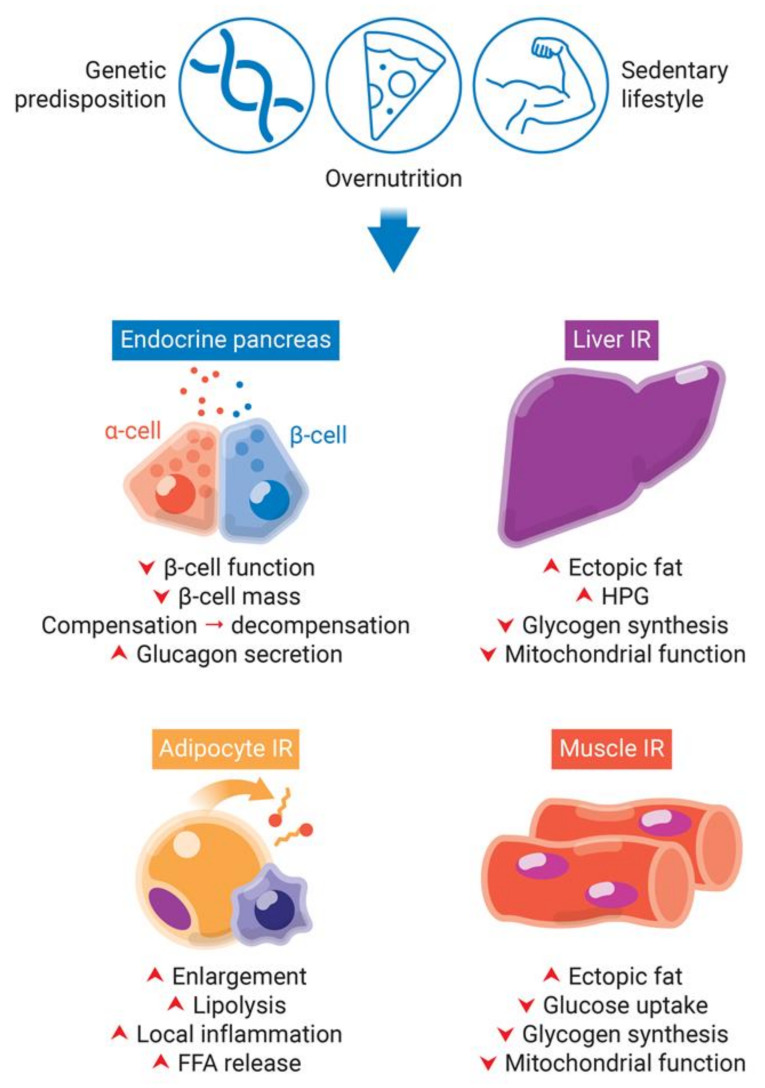
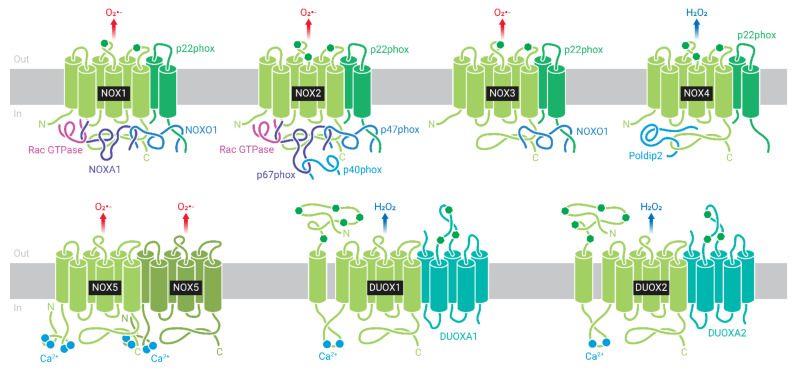
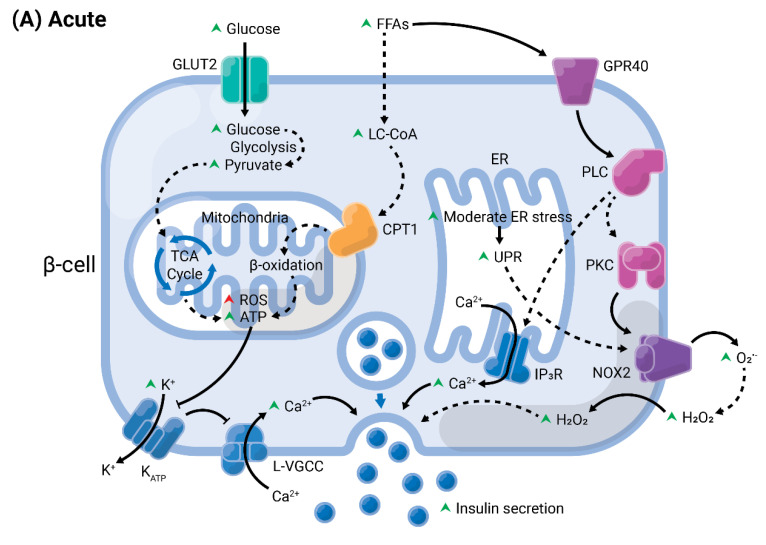
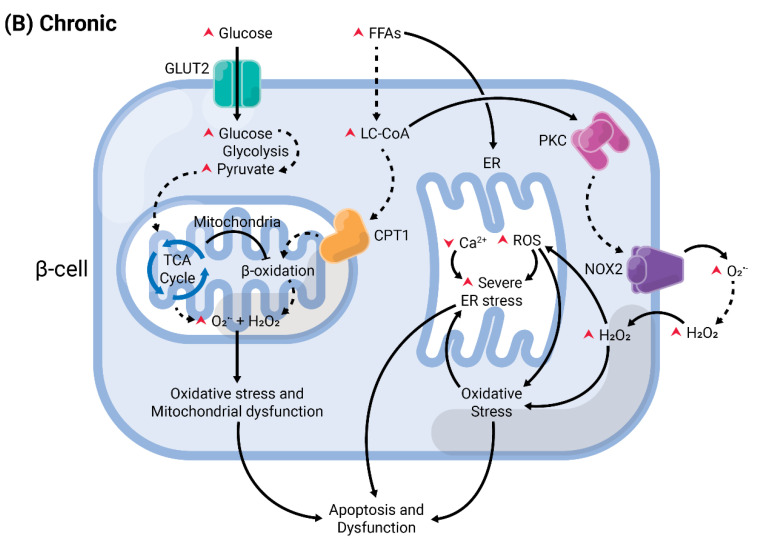
A high caloric intake, rich in saturated fats, greatly contributes to the development of obesity, which is the leading risk factor for type 2 diabetes (T2D). A persistent caloric surplus increases plasma levels of fatty acids (FAs), especially saturated ones, which were shown to negatively impact pancreatic β-cell function and survival in a process called lipotoxicity. Lipotoxicity in β-cells activates different stress pathways, culminating in β-cells dysfunction and death. Among all stresses, endoplasmic reticulum (ER) stress and oxidative stress have been shown to be strongly correlated. One main source of oxidative stress in pancreatic β-cells appears to be the reactive oxygen species producer NADPH oxidase (NOX) enzyme, which has a role in the glucose-stimulated insulin secretion and in the β-cell demise during both T1 and T2D. In this review, we focus on the acute and chronic effects of FAs and the lipotoxicity-induced β-cell failure during T2D development, with special emphasis on the oxidative stress induced by NOX, the ER stress, and the crosstalk between NOX and ER stress.
Keywords: ER stress; NADPH oxidase; lipotoxicity; oxidative stress; pancreatic β-cell; type 2 diabetes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction.Cells. 2021 Jun 22;10(7):1573. doi: 10.3390/cells10071573. Cells. 2021. PMID: 34206537 Free PMC article. Review.
-
Metformin Ameliorates Lipotoxic β-Cell Dysfunction through a Concentration-Dependent Dual Mechanism of Action.Diabetes Metab J. 2019 Dec;43(6):854-866. doi: 10.4093/dmj.2018.0179. Epub 2019 Jun 27. Diabetes Metab J. 2019. PMID: 31339010 Free PMC article.
-
Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells.Redox Biol. 2017 Apr;11:126-134. doi: 10.1016/j.redox.2016.11.009. Epub 2016 Nov 23. Redox Biol. 2017. PMID: 27912197 Free PMC article.
-
Myricetin prevents thapsigargin-induced CDK5-P66Shc signalosome mediated pancreatic β-cell dysfunction.Free Radic Biol Med. 2019 Sep;141:59-66. doi: 10.1016/j.freeradbiomed.2019.05.038. Epub 2019 Jun 1. Free Radic Biol Med. 2019. PMID: 31163256
-
Molecular mechanisms of lipotoxicity-induced pancreatic β-cell dysfunction.Int Rev Cell Mol Biol. 2021;359:357-402. doi: 10.1016/bs.ircmb.2021.02.013. Epub 2021 Mar 12. Int Rev Cell Mol Biol. 2021. PMID: 33832653 Review.
Cited by
-
Stem Cell-Derived Islets for Type 2 Diabetes.Int J Mol Sci. 2022 May 4;23(9):5099. doi: 10.3390/ijms23095099. Int J Mol Sci. 2022. PMID: 35563490 Free PMC article. Review.
-
Effects of dulaglutide and trelagliptin on beta-cell function in patients with type 2 diabetes: a randomized controlled study: DUET-beta study.Diabetol Int. 2024 Apr 3;15(3):474-482. doi: 10.1007/s13340-024-00717-6. eCollection 2024 Jul. Diabetol Int. 2024. PMID: 39101164
-
Effect of metformin on Wnt5a in individuals new-onset type 2 diabetes with different body mass indexes: The evidences from the real word research.J Diabetes Metab Disord. 2023 Aug 26;22(2):1561-1570. doi: 10.1007/s40200-023-01286-2. eCollection 2023 Dec. J Diabetes Metab Disord. 2023. PMID: 37975126 Free PMC article.
-
Ameliorative Effects of Curcumin on Type 2 Diabetes Mellitus.Molecules. 2024 Jun 20;29(12):2934. doi: 10.3390/molecules29122934. Molecules. 2024. PMID: 38930998 Free PMC article. Review.
-
Transcriptomic analysis reveals the crosstalk between type 2 diabetes and chronic pancreatitis.Health Sci Rep. 2024 Apr 29;7(5):e2079. doi: 10.1002/hsr2.2079. eCollection 2024 May. Health Sci Rep. 2024. PMID: 38690006 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
