

Chemical Reporters for Bacterial Glycans: Development and Applications
- PMID: 34905344
- PMCID: PMC8958928
- DOI: 10.1021/acs.chemrev.1c00729
Chemical Reporters for Bacterial Glycans: Development and Applications
Abstract
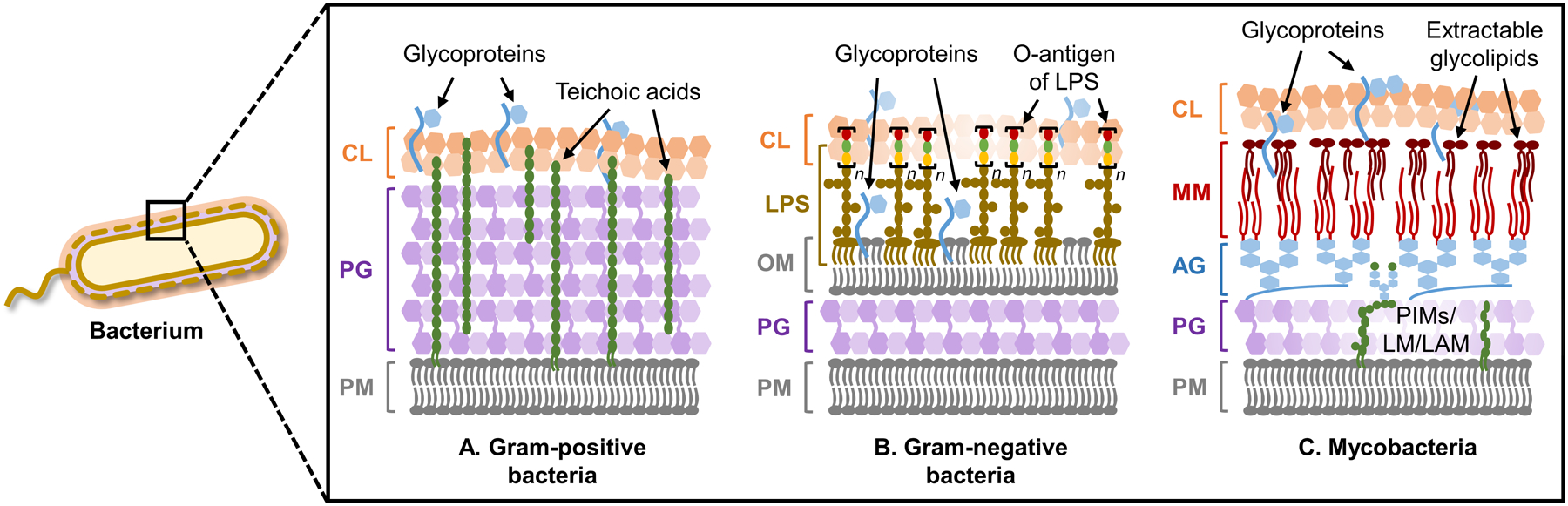
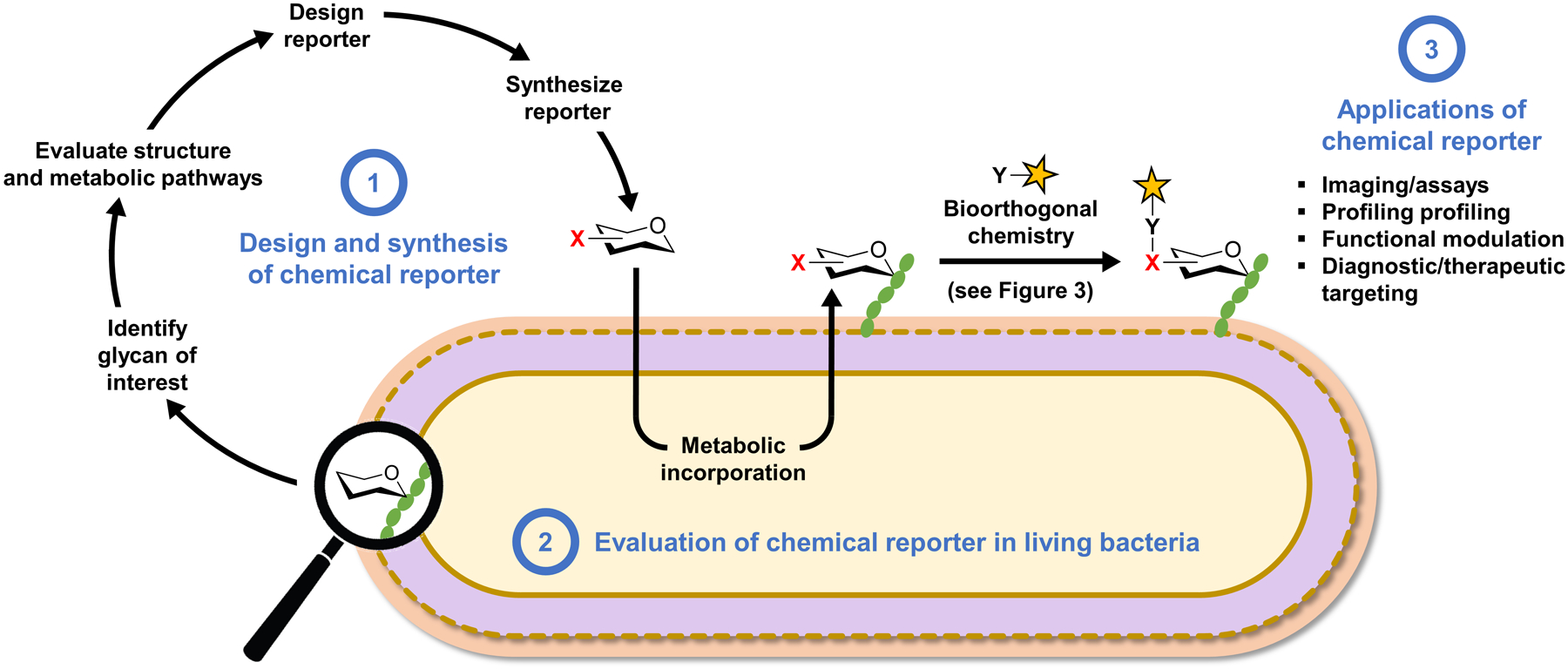
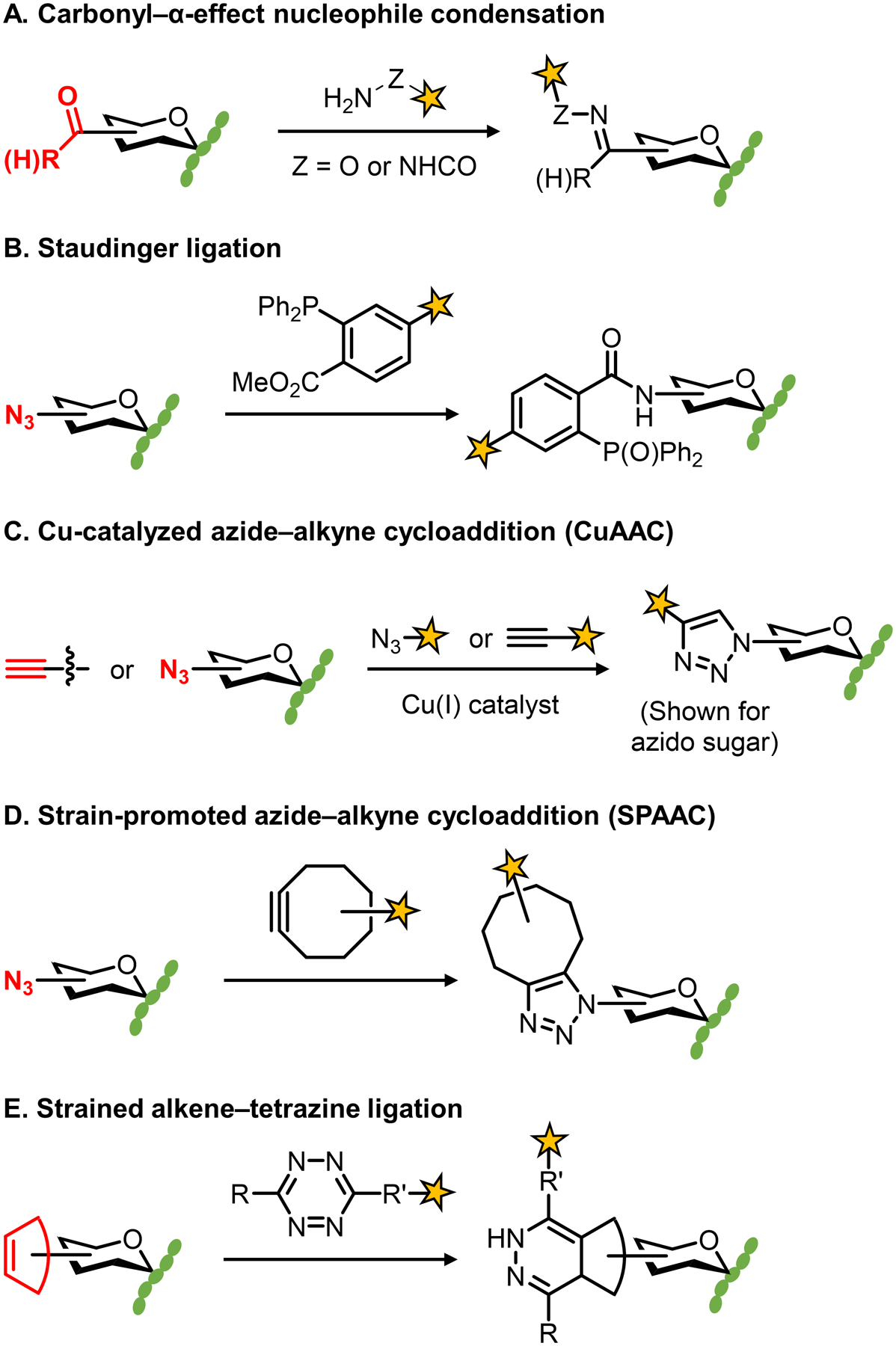
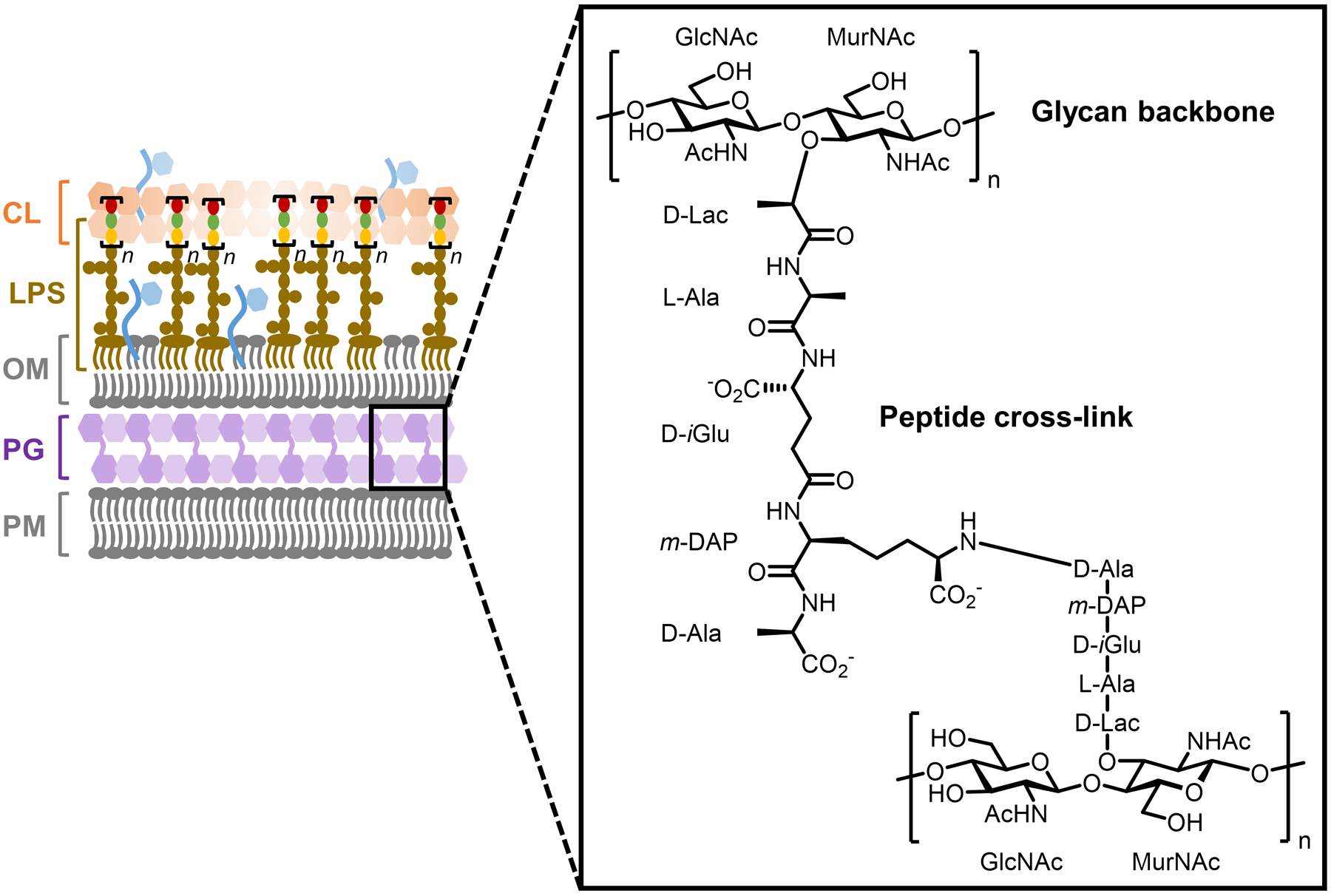
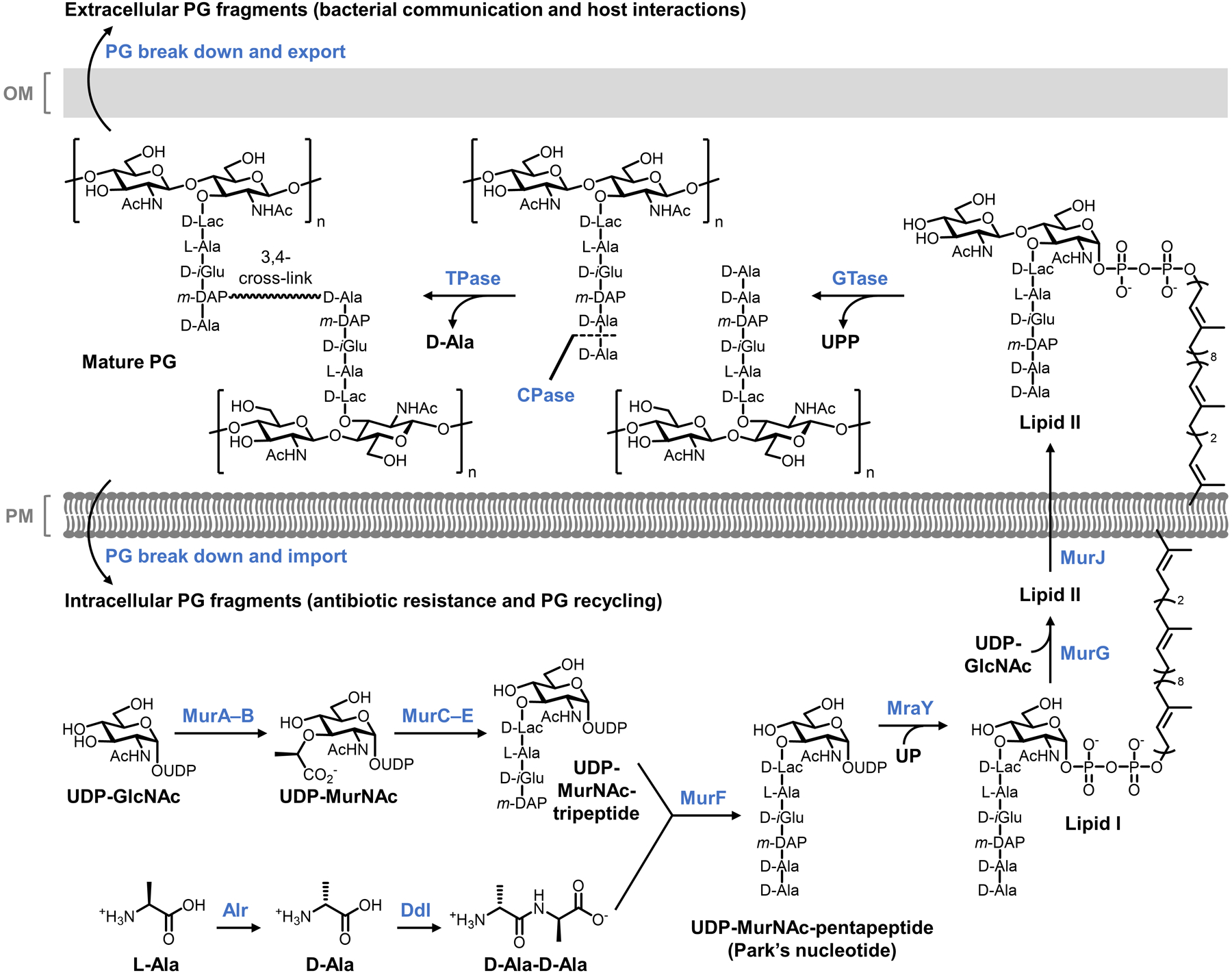
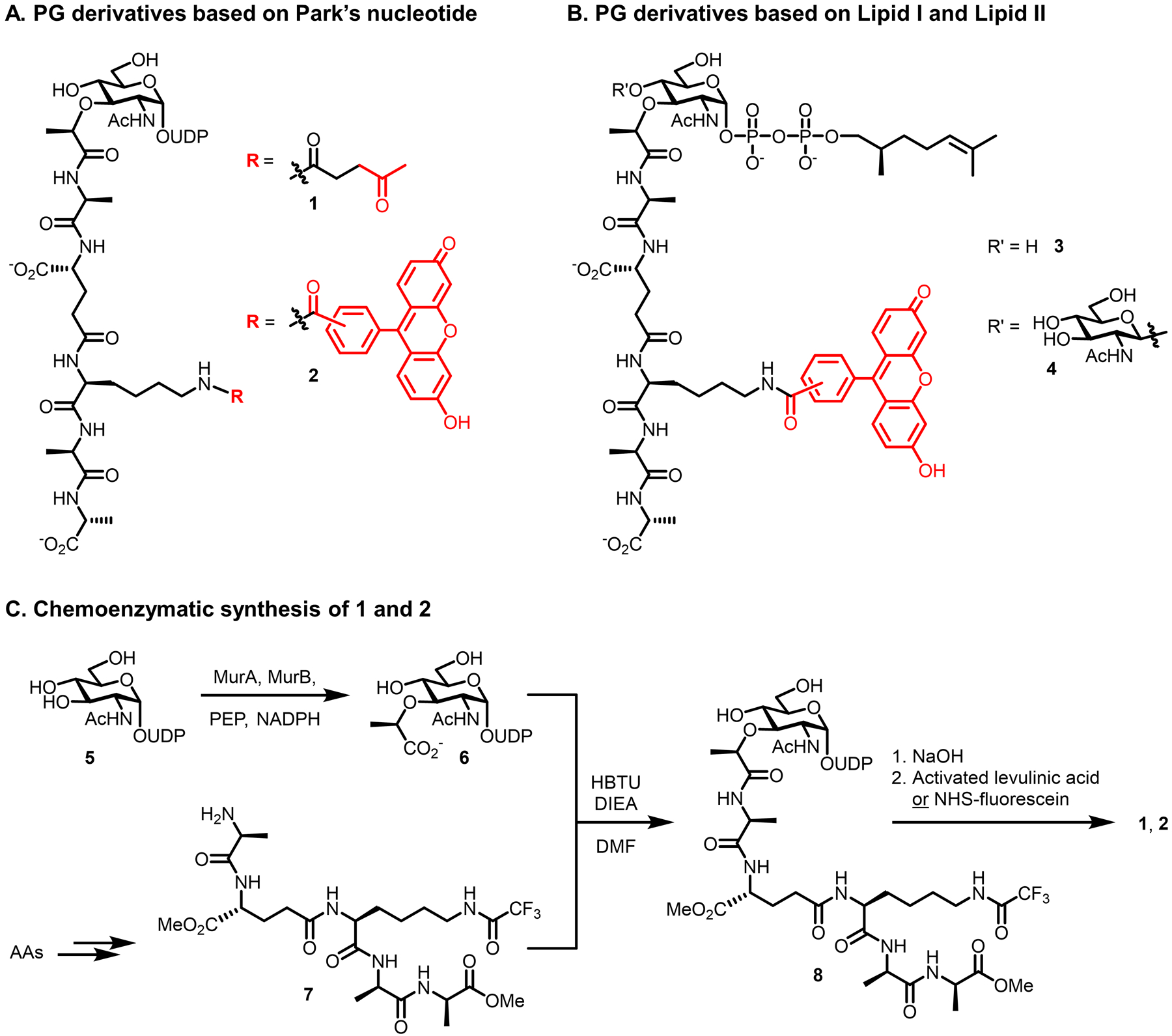
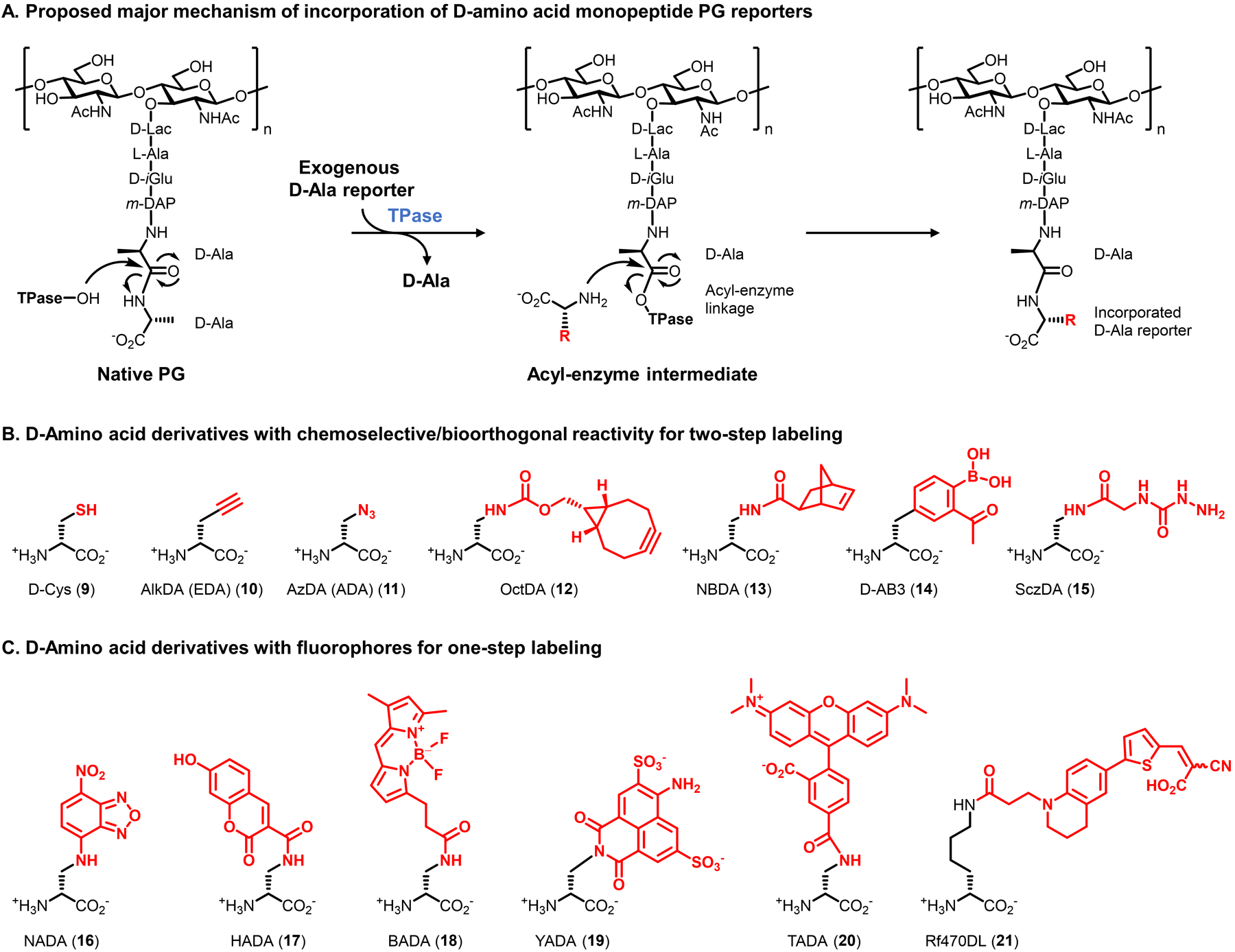
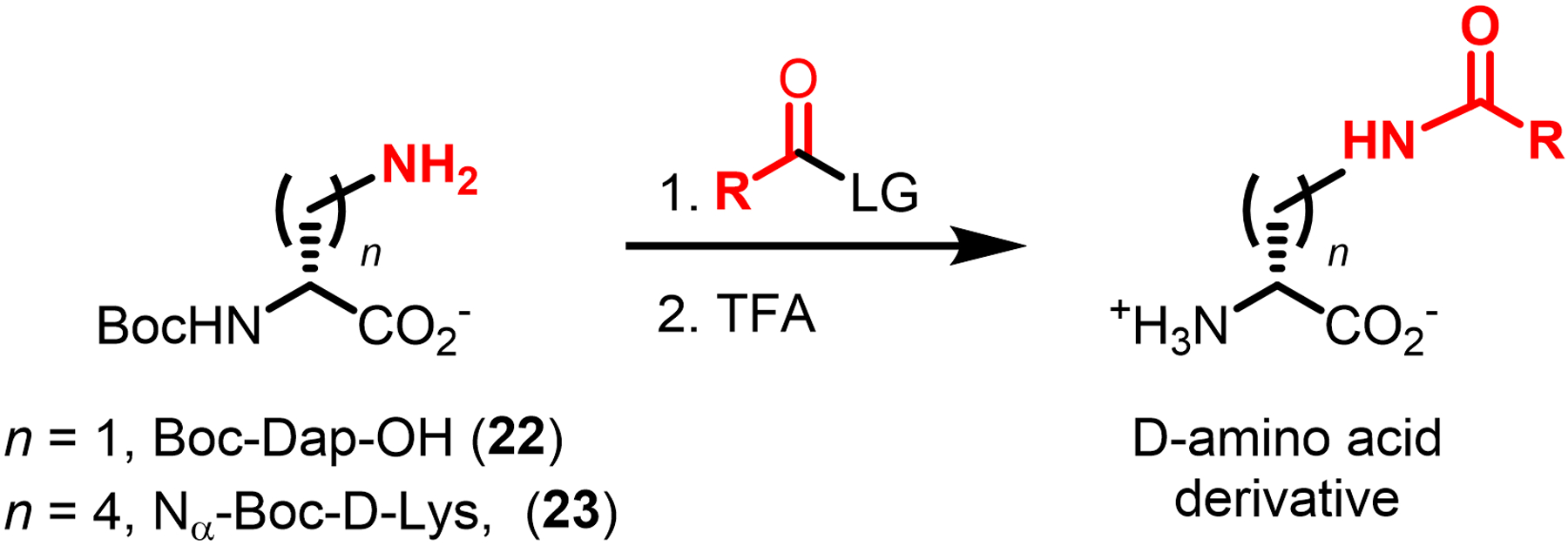
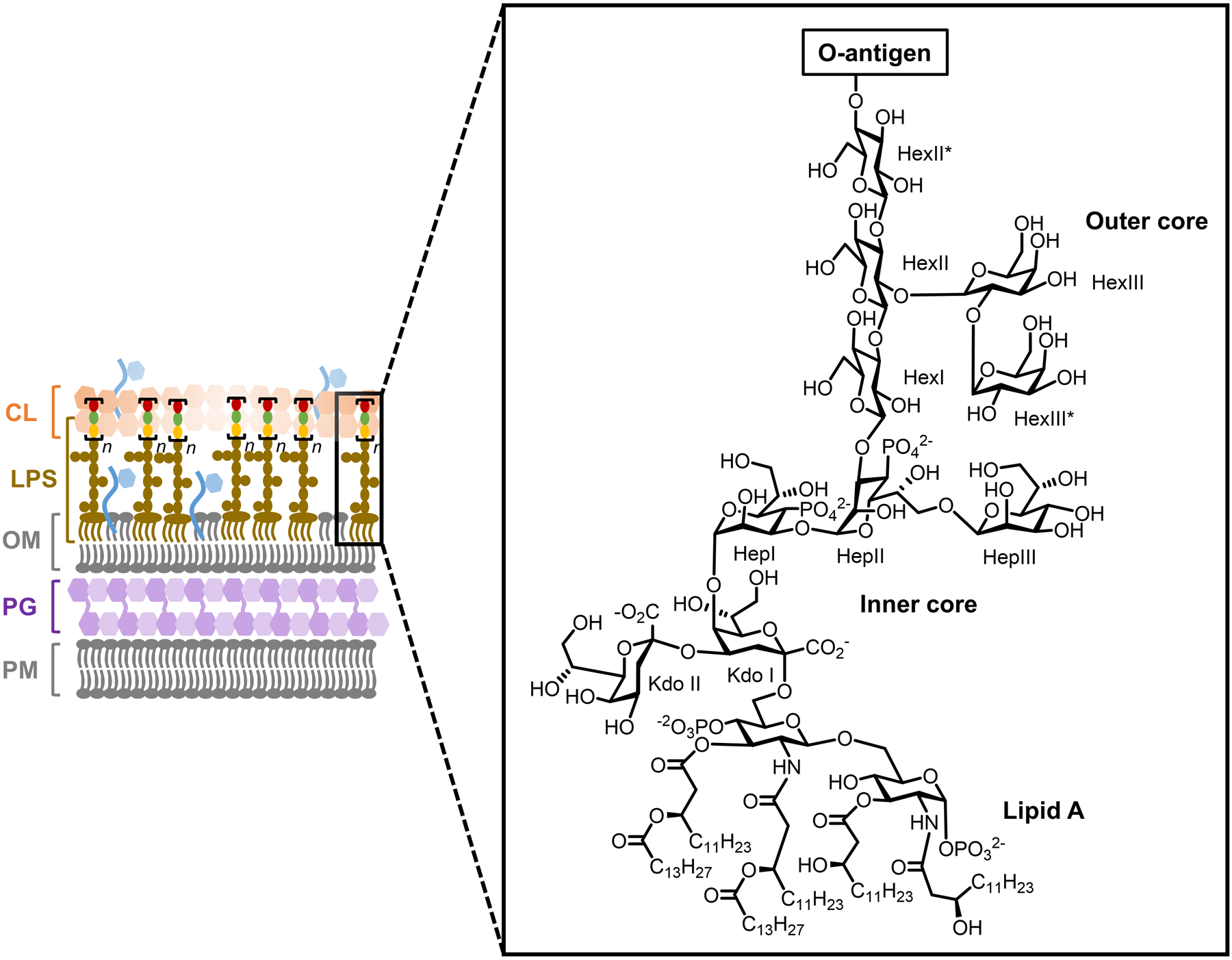
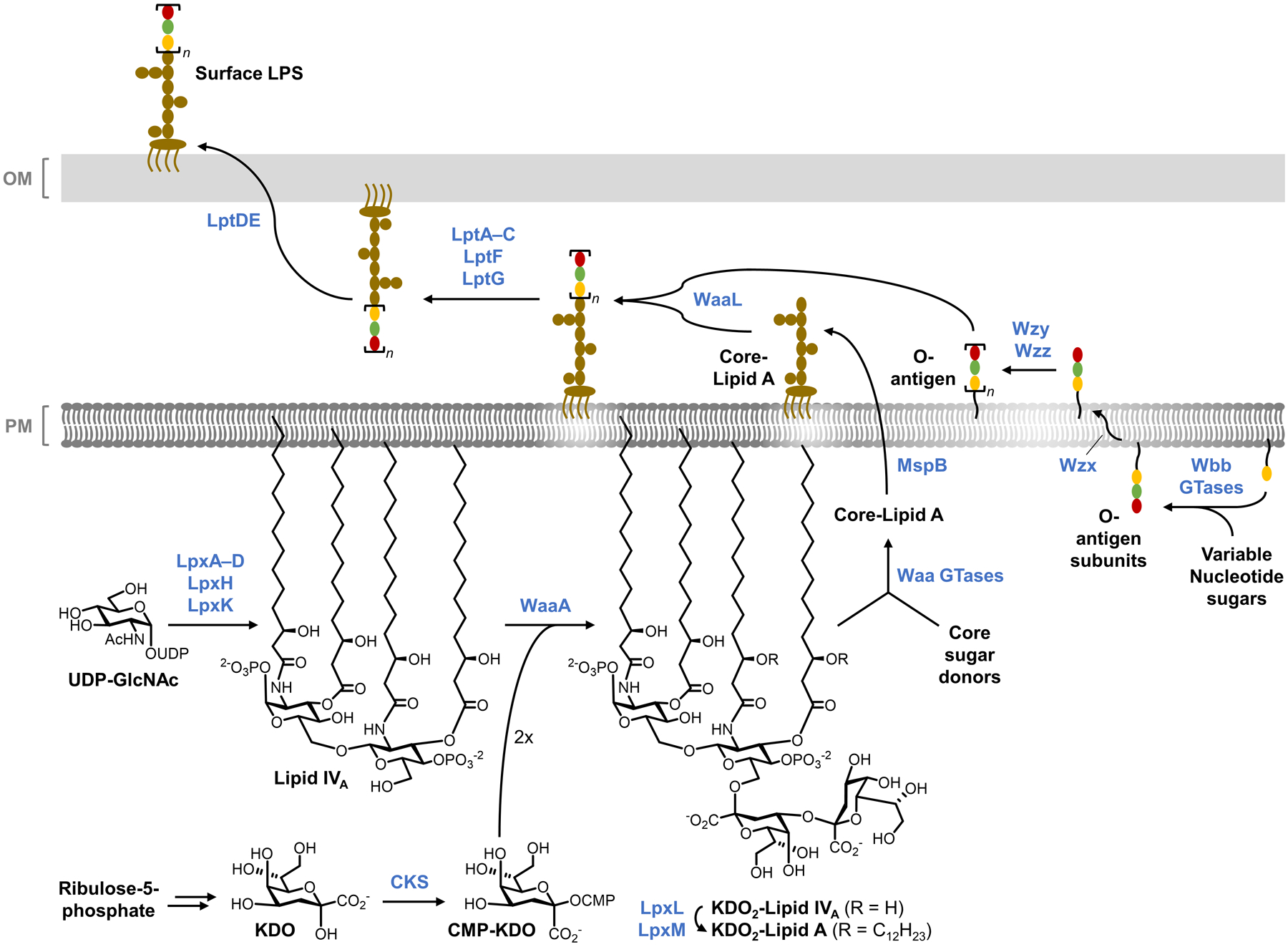
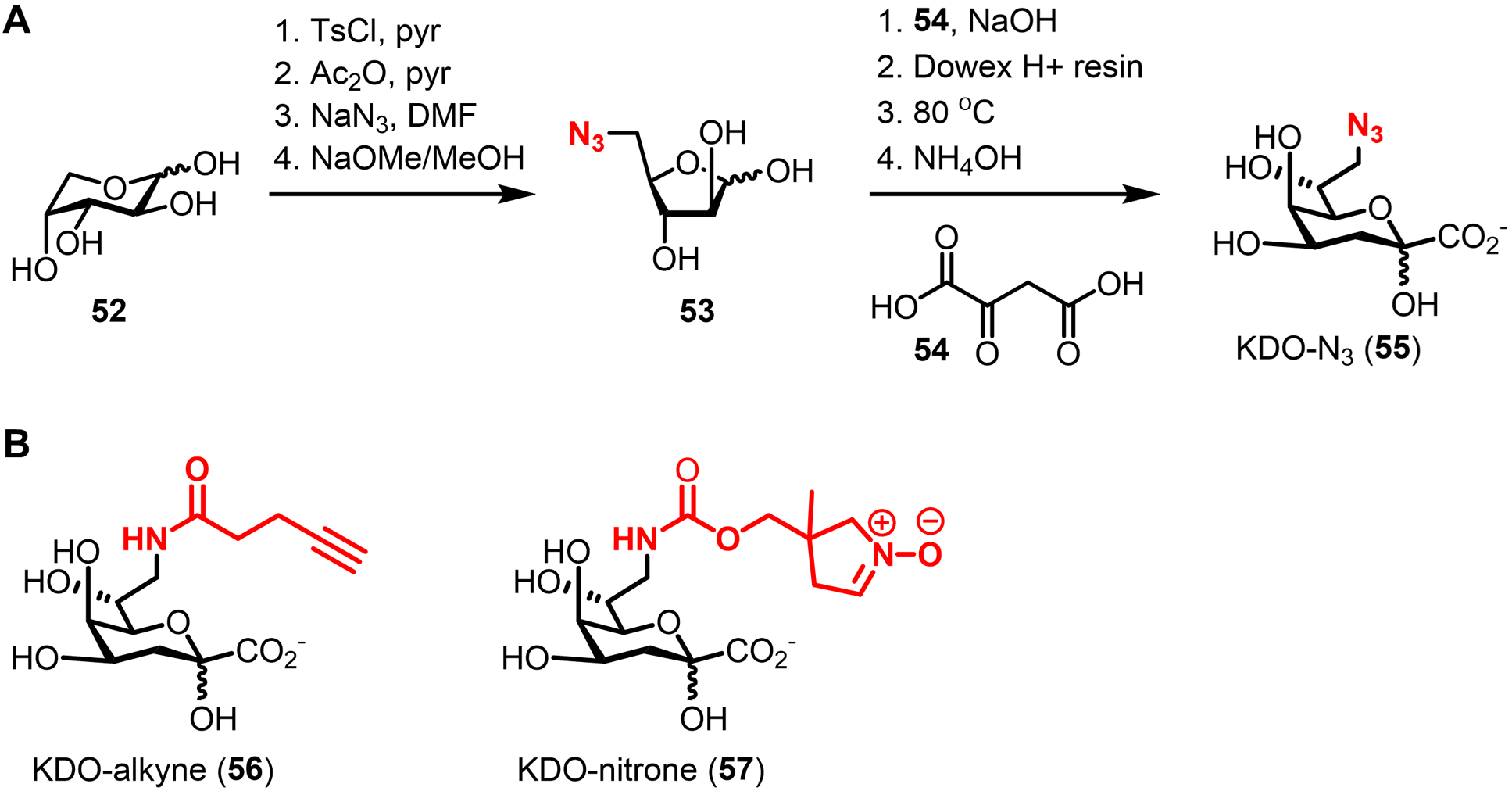
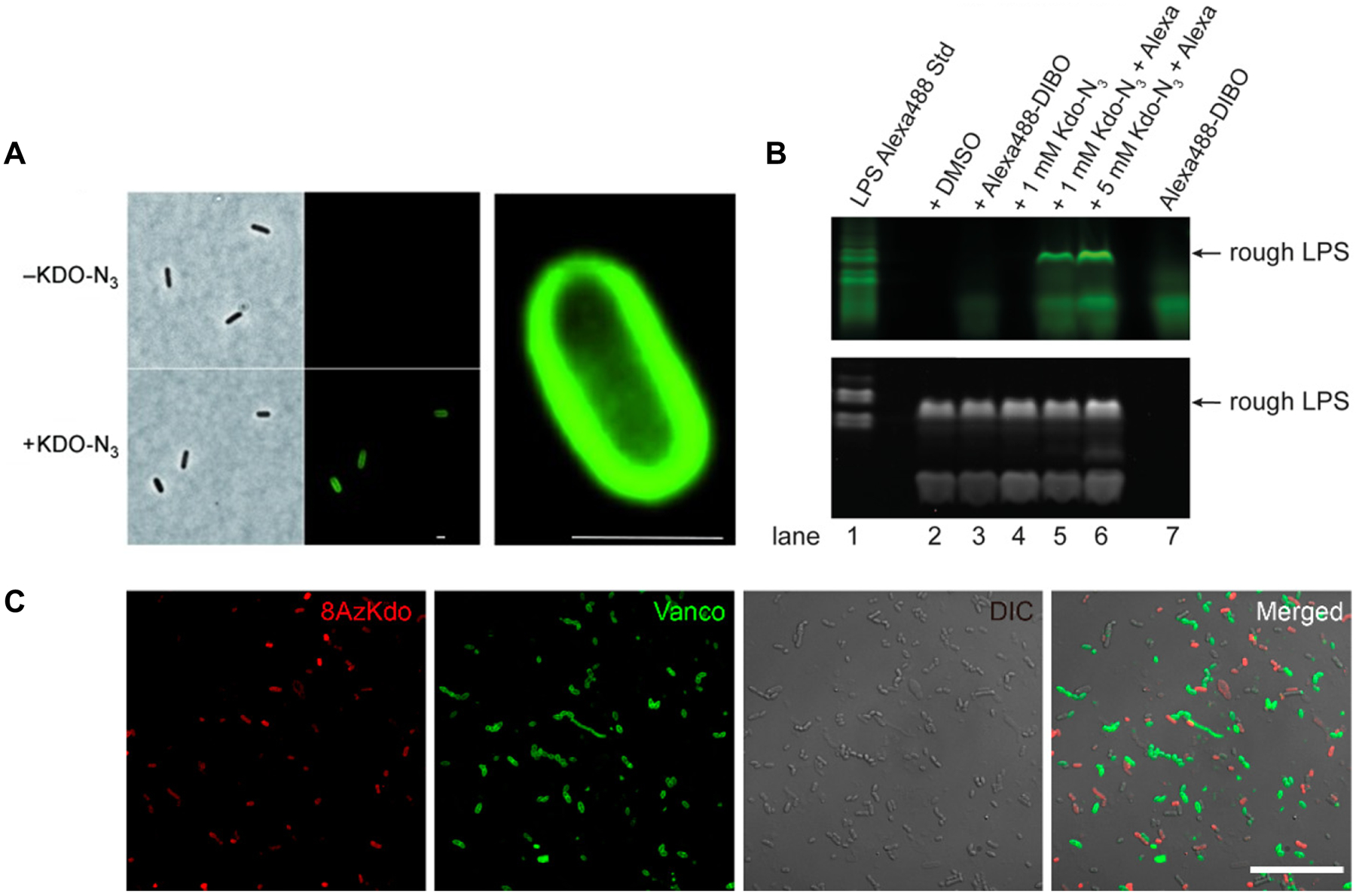
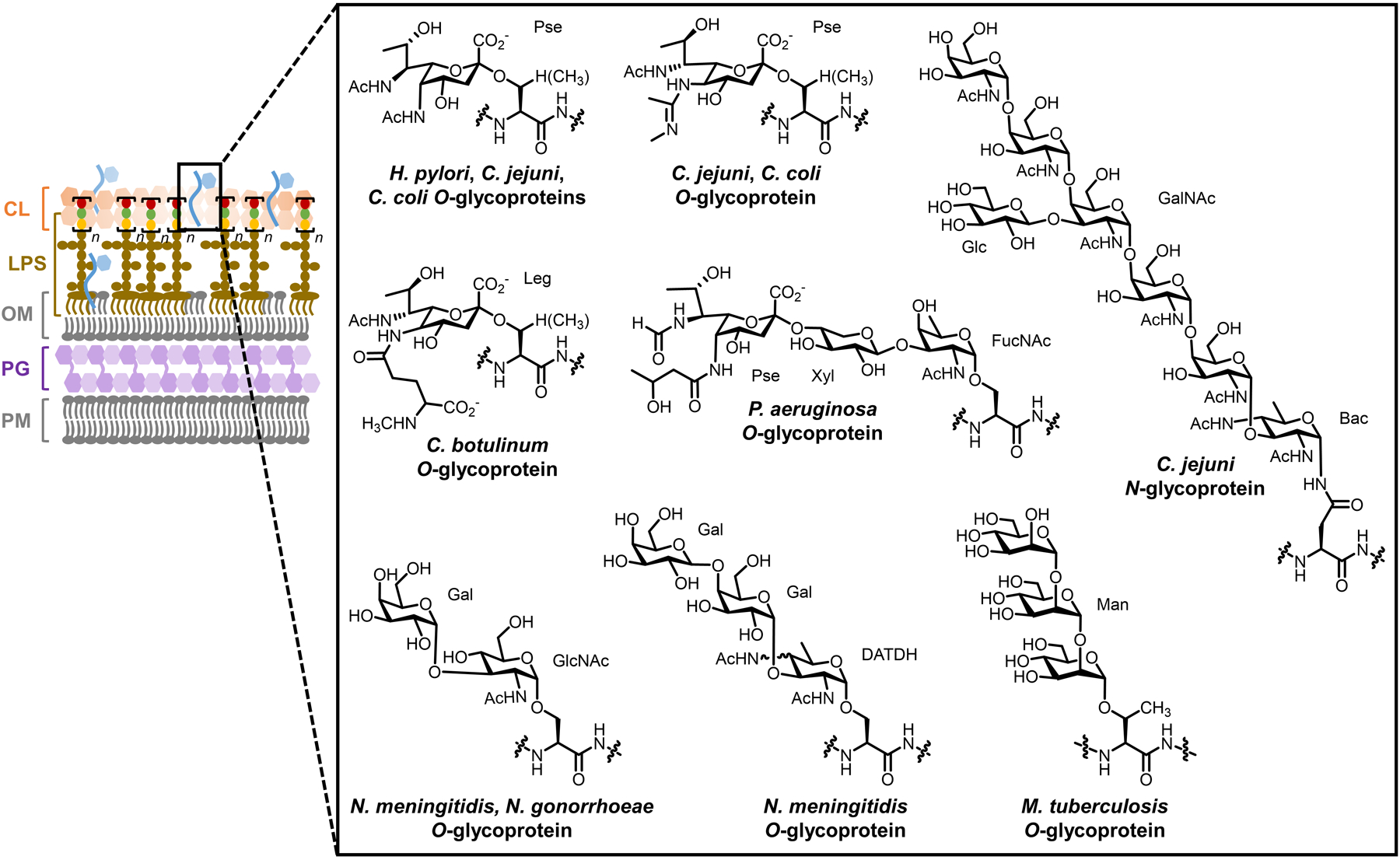
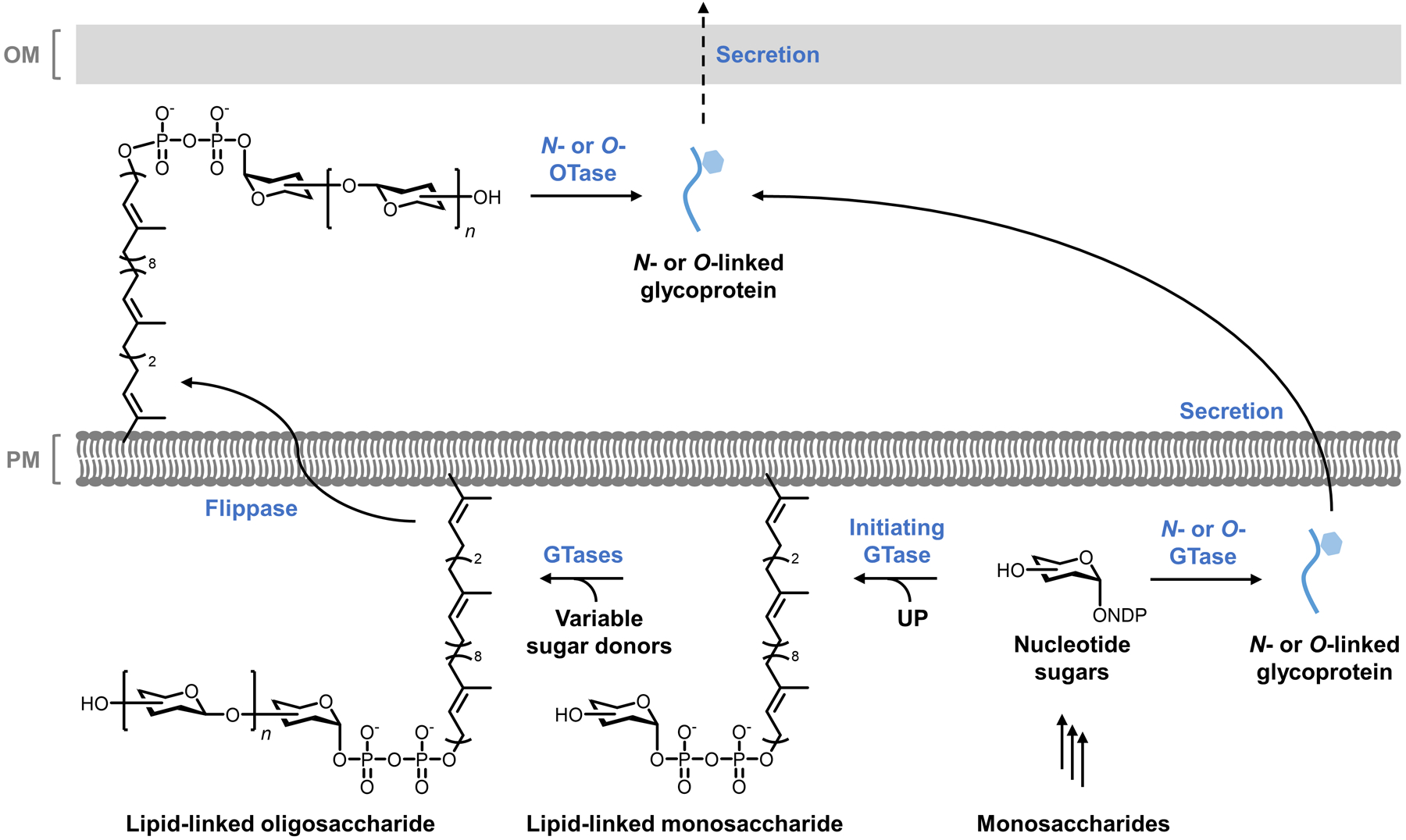
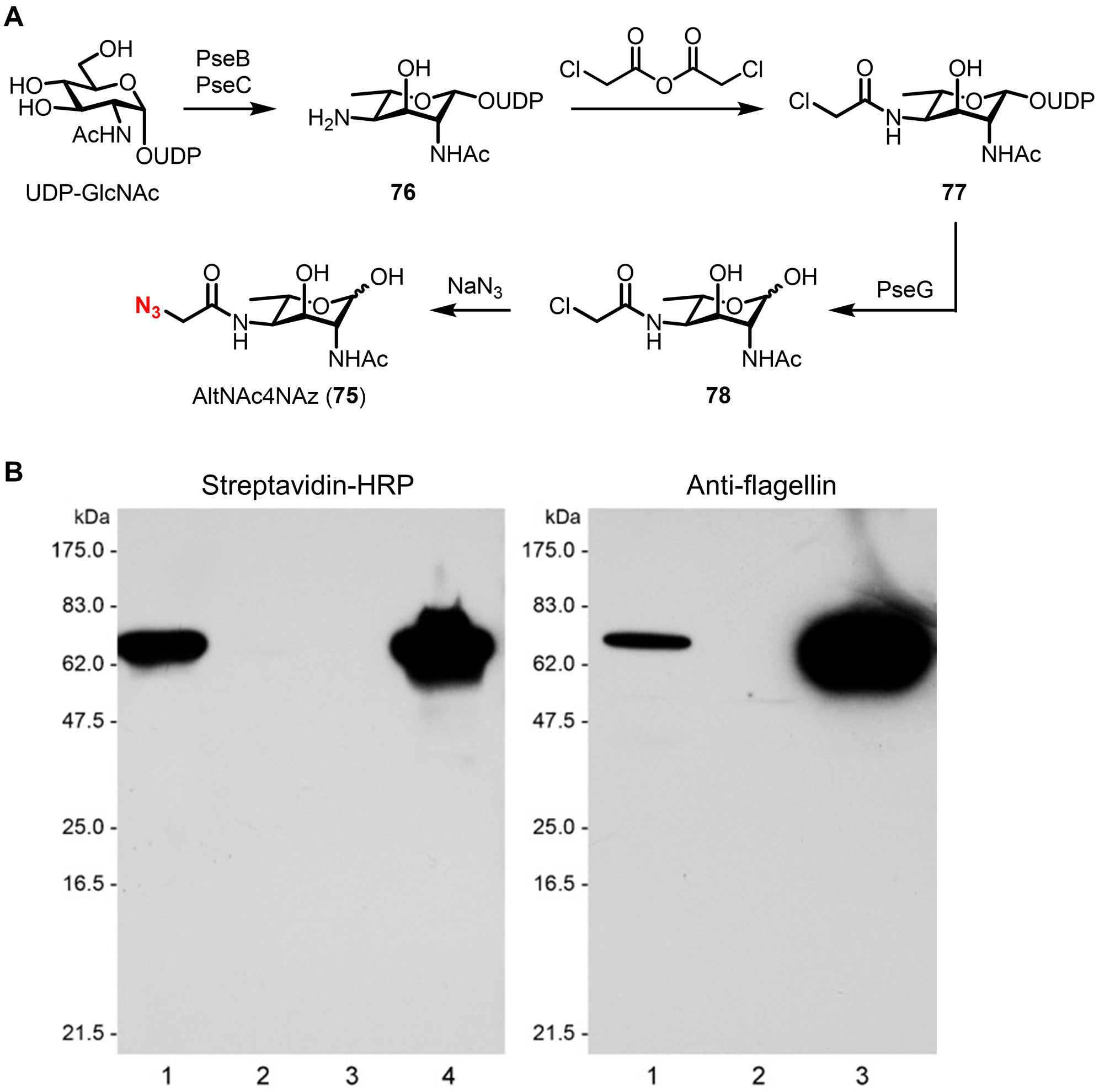
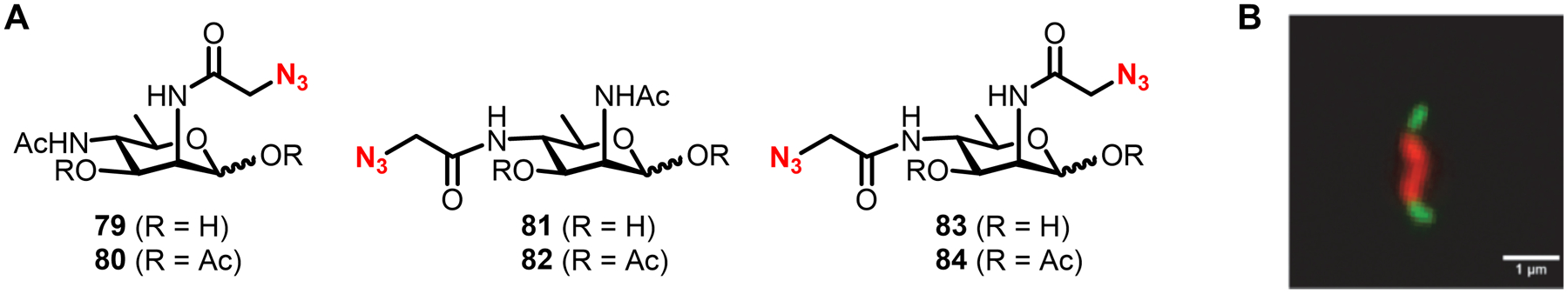
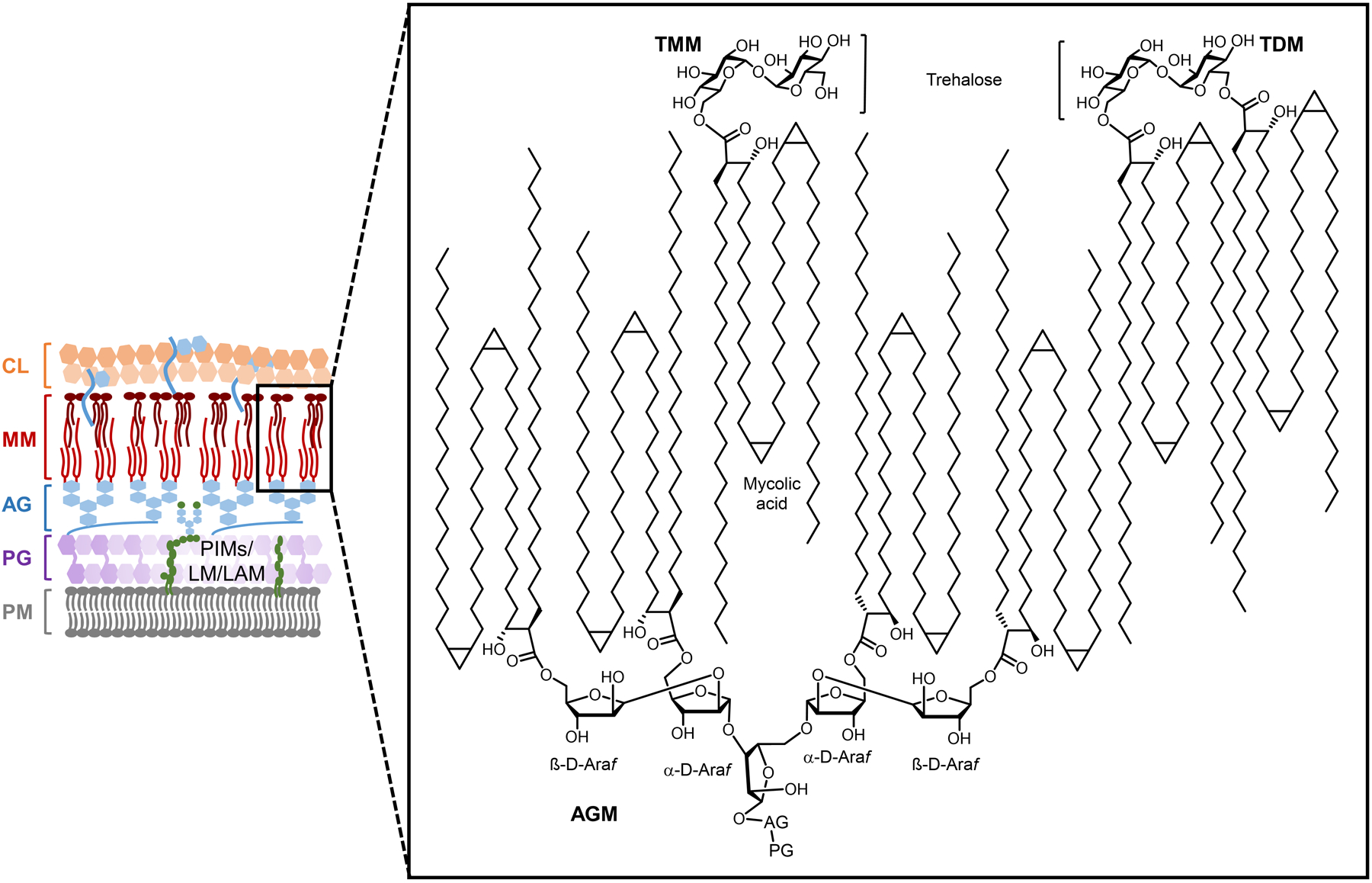
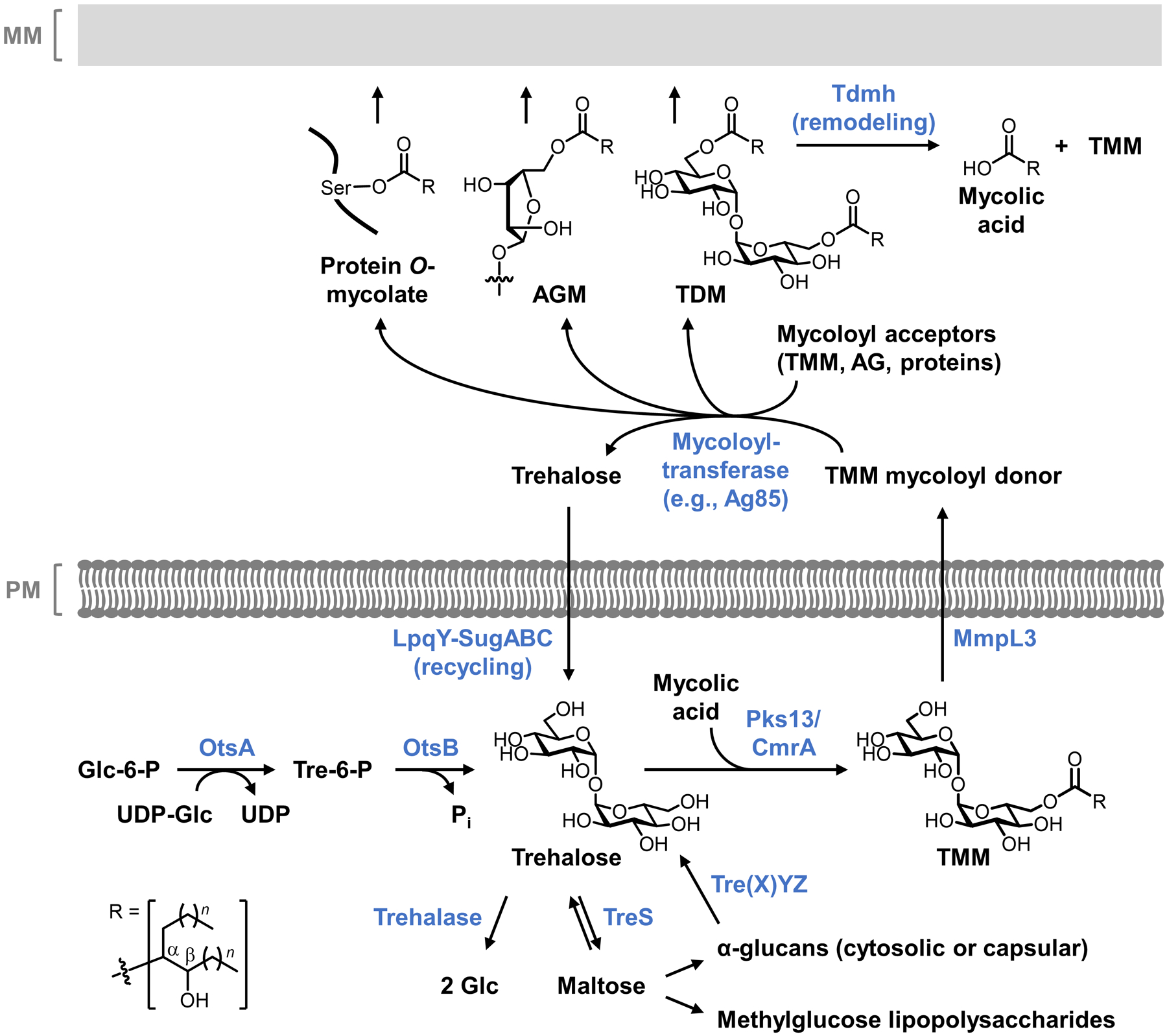
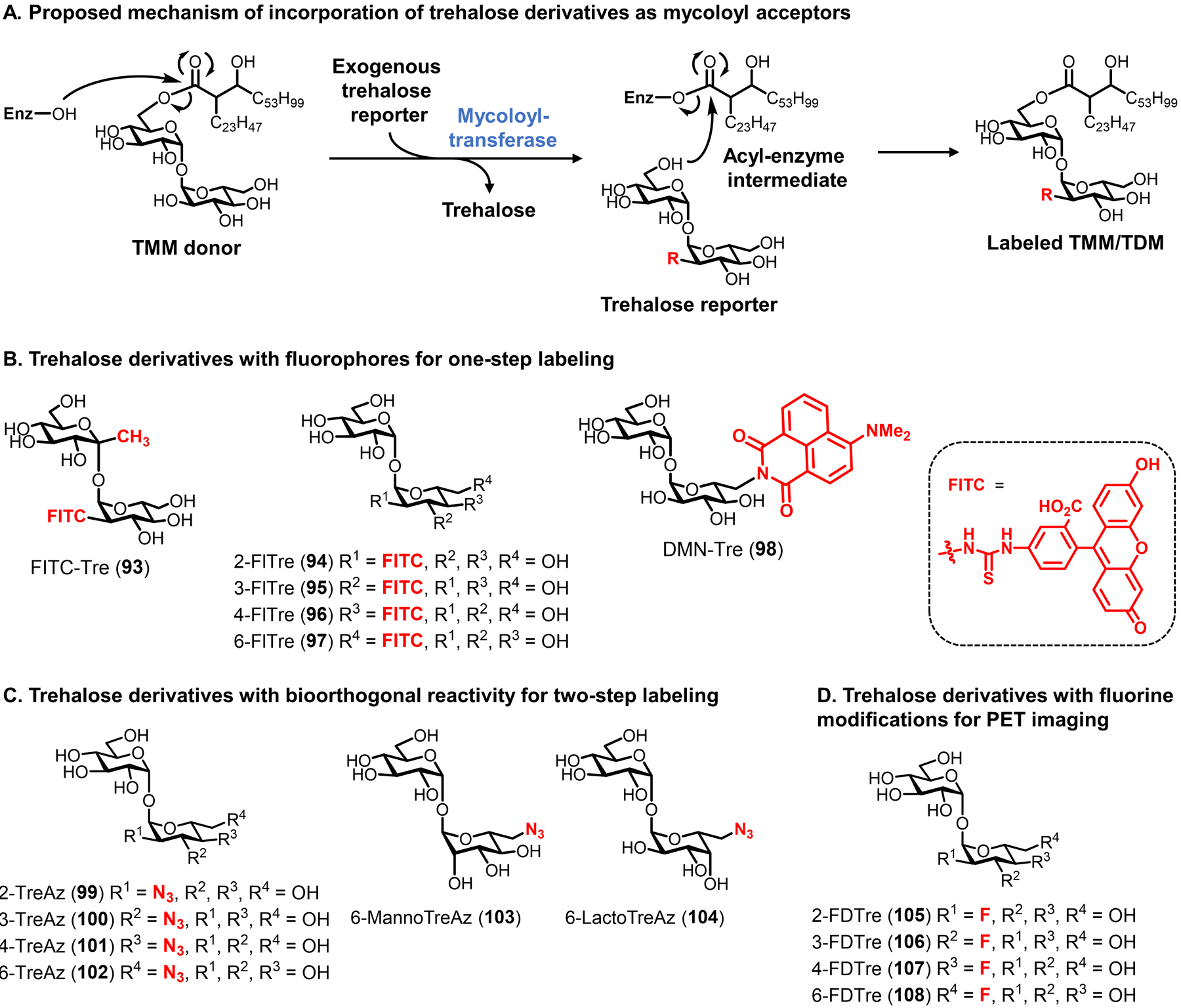
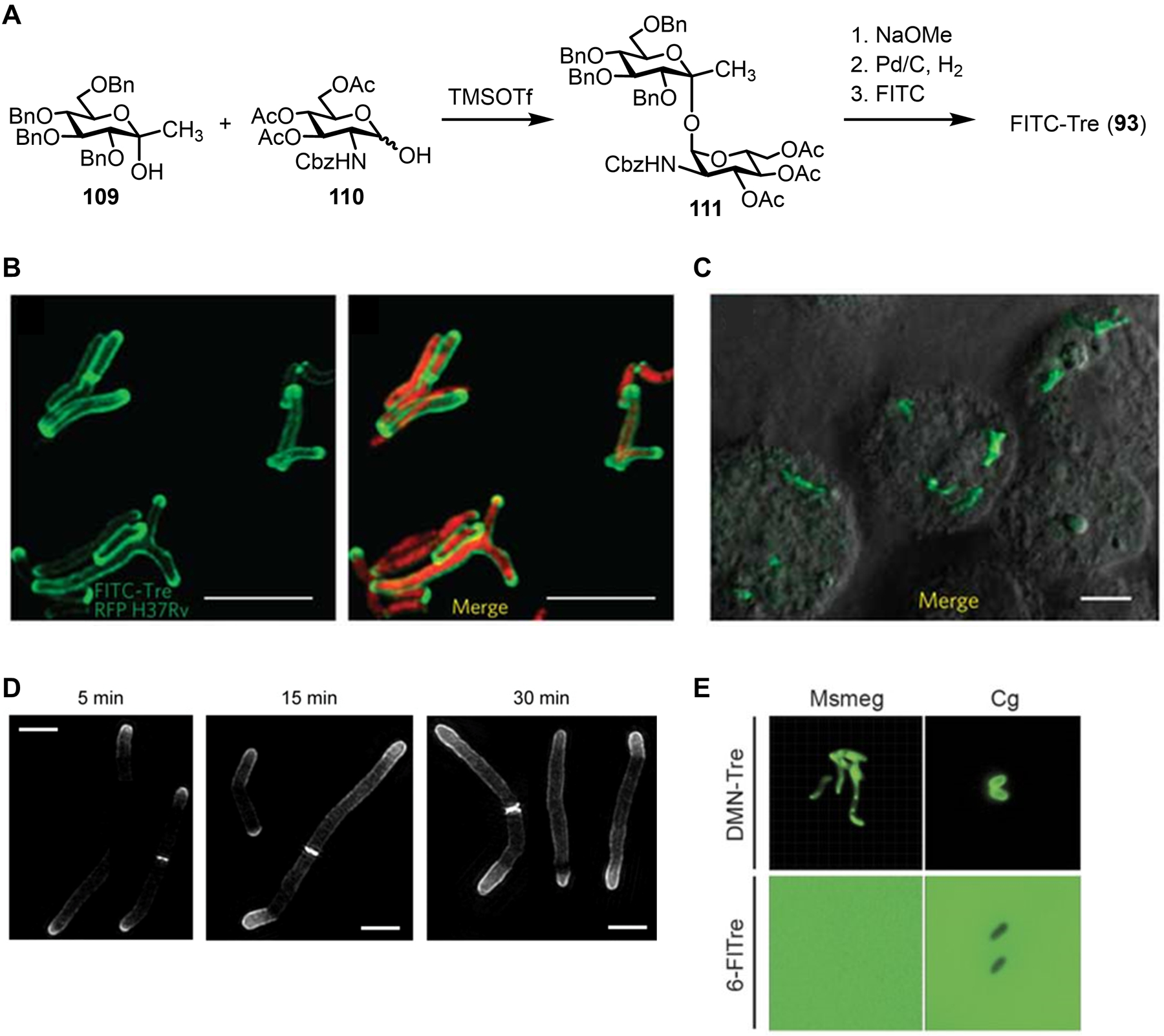
Bacteria possess an extraordinary repertoire of cell envelope glycans that have critical physiological functions. Pathogenic bacteria have glycans that are essential for growth and virulence but are absent from humans, making them high-priority targets for antibiotic, vaccine, and diagnostic development. The advent of metabolic labeling with bioorthogonal chemical reporters and small-molecule fluorescent reporters has enabled the investigation and targeting of specific bacterial glycans in their native environments. These tools have opened the door to imaging glycan dynamics, assaying and inhibiting glycan biosynthesis, profiling glycoproteins and glycan-binding proteins, and targeting pathogens with diagnostic and therapeutic payload. These capabilities have been wielded in diverse commensal and pathogenic Gram-positive, Gram-negative, and mycobacterial species─including within live host organisms. Here, we review the development and applications of chemical reporters for bacterial glycans, including peptidoglycan, lipopolysaccharide, glycoproteins, teichoic acids, and capsular polysaccharides, as well as mycobacterial glycans, including trehalose glycolipids and arabinan-containing glycoconjugates. We cover in detail how bacteria-targeting chemical reporters are designed, synthesized, and evaluated, how they operate from a mechanistic standpoint, and how this information informs their judicious and innovative application. We also provide a perspective on the current state and future directions of the field, underscoring the need for interdisciplinary teams to create novel tools and extend existing tools to support fundamental and translational research on bacterial glycans.
Figures
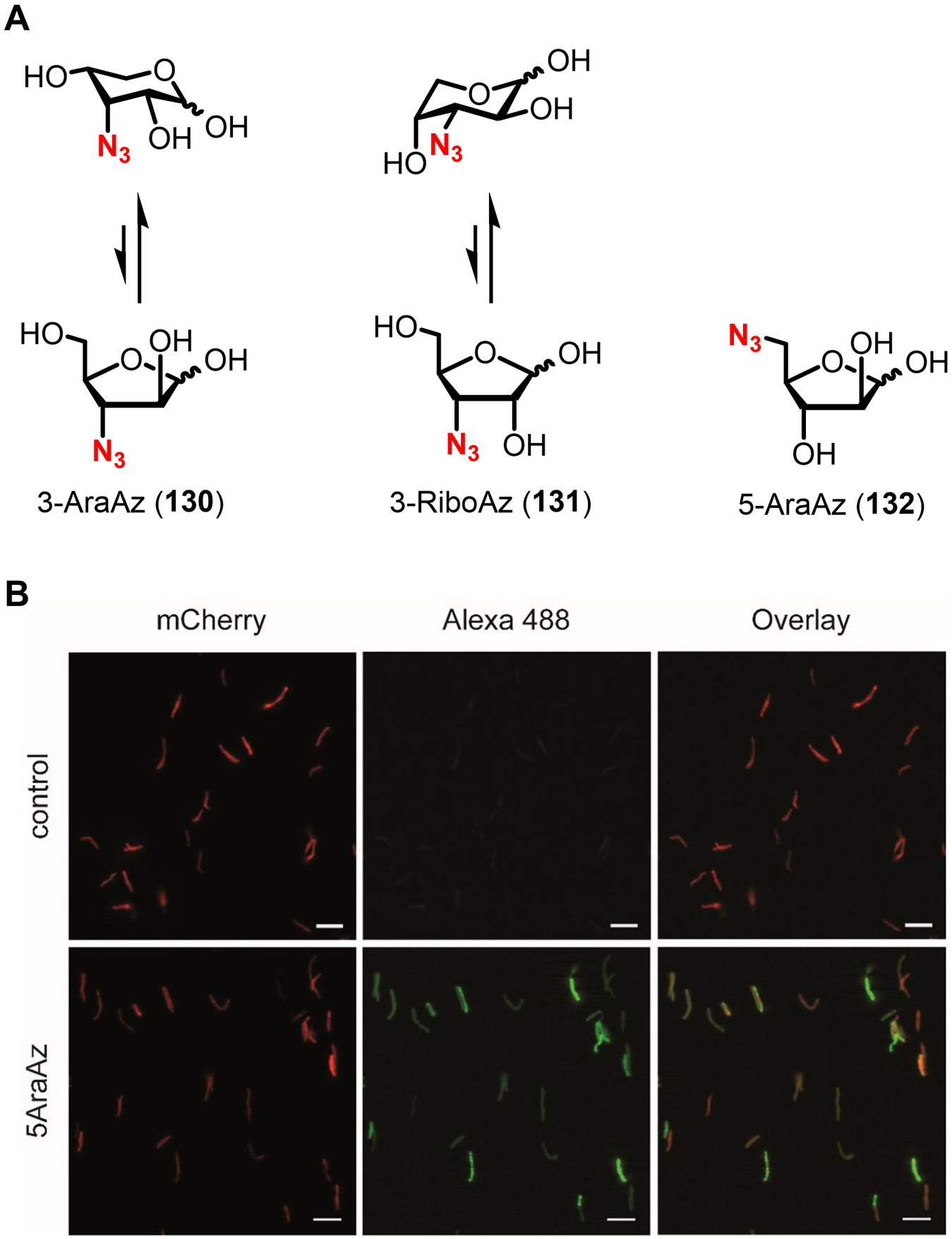
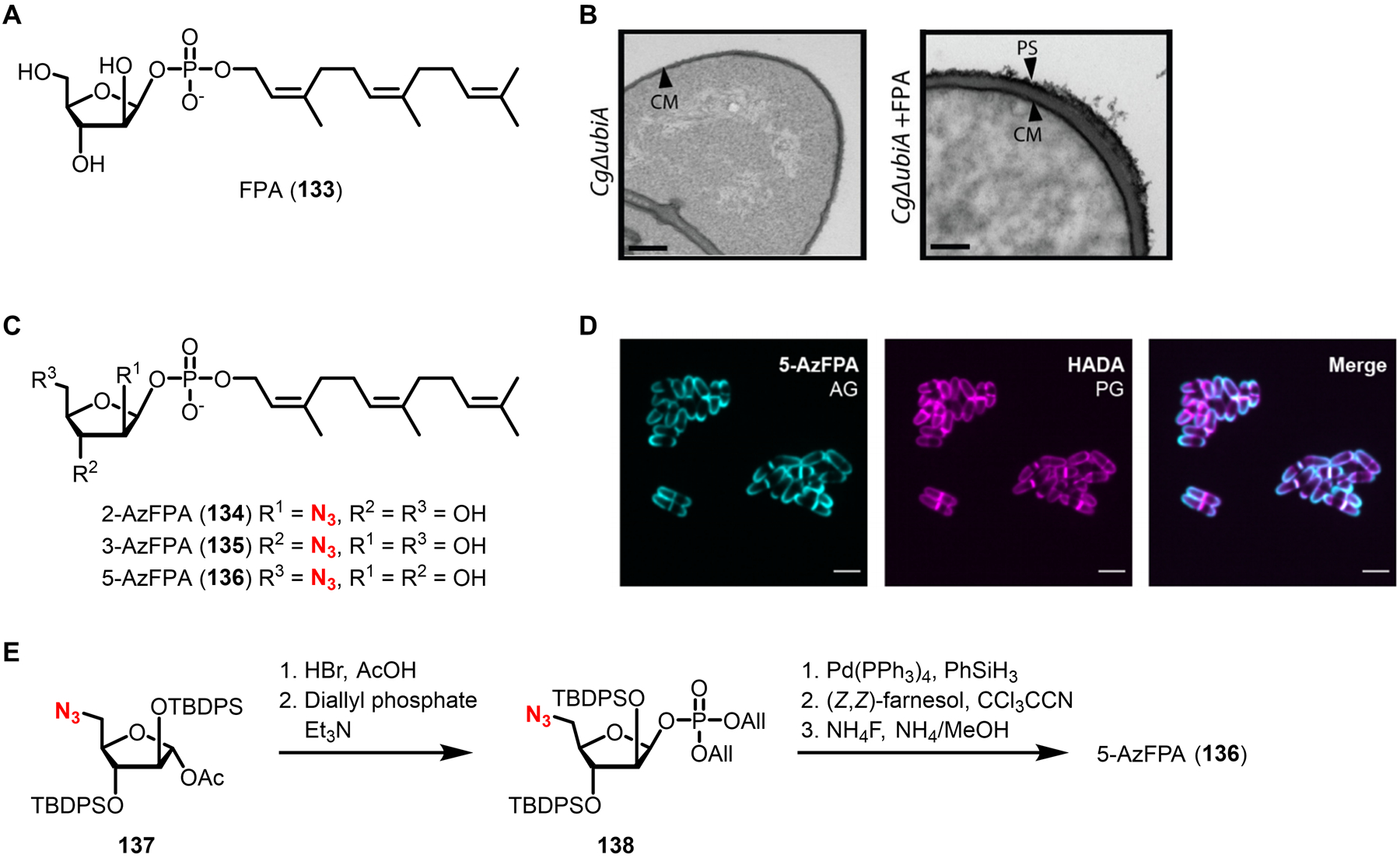
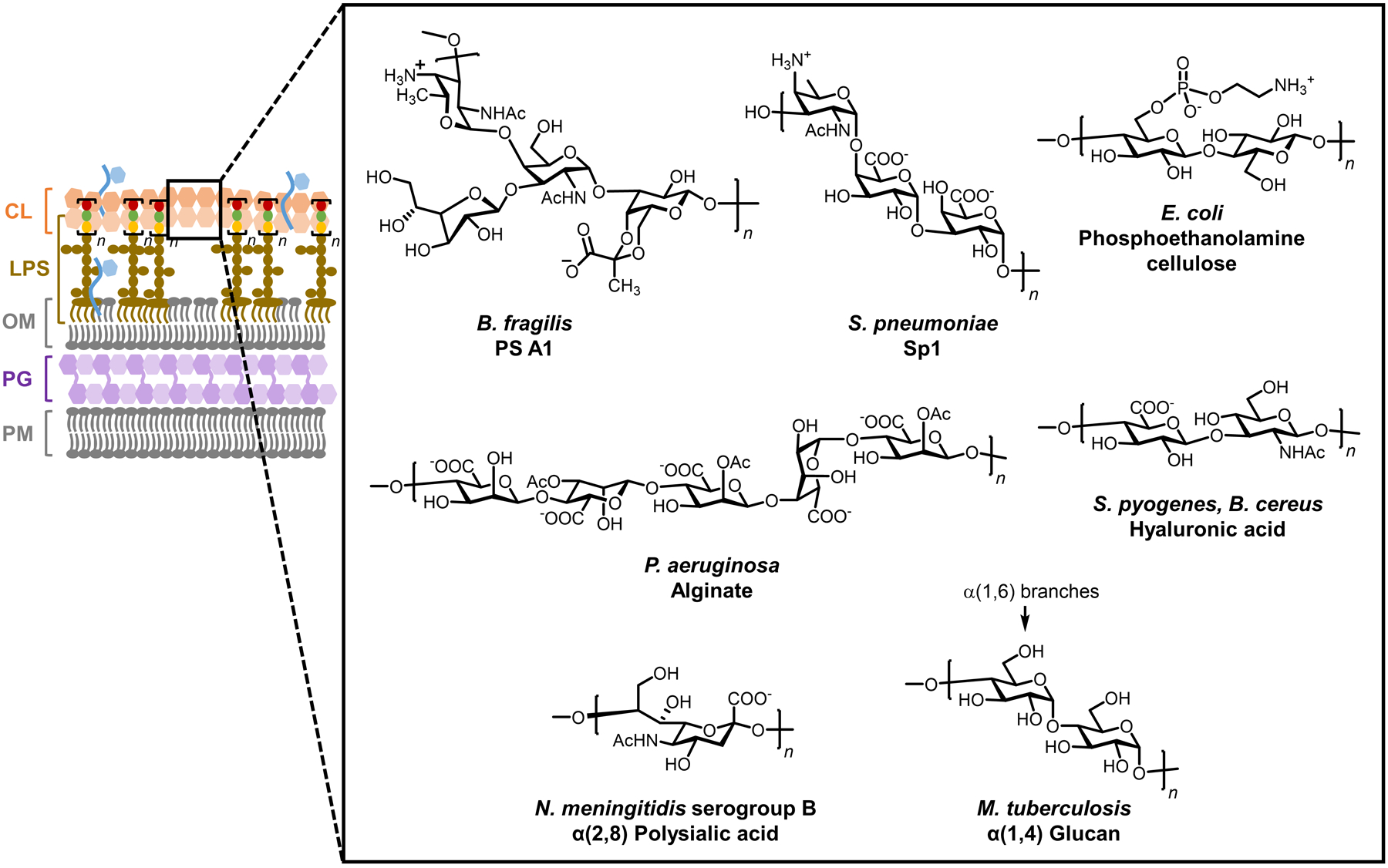
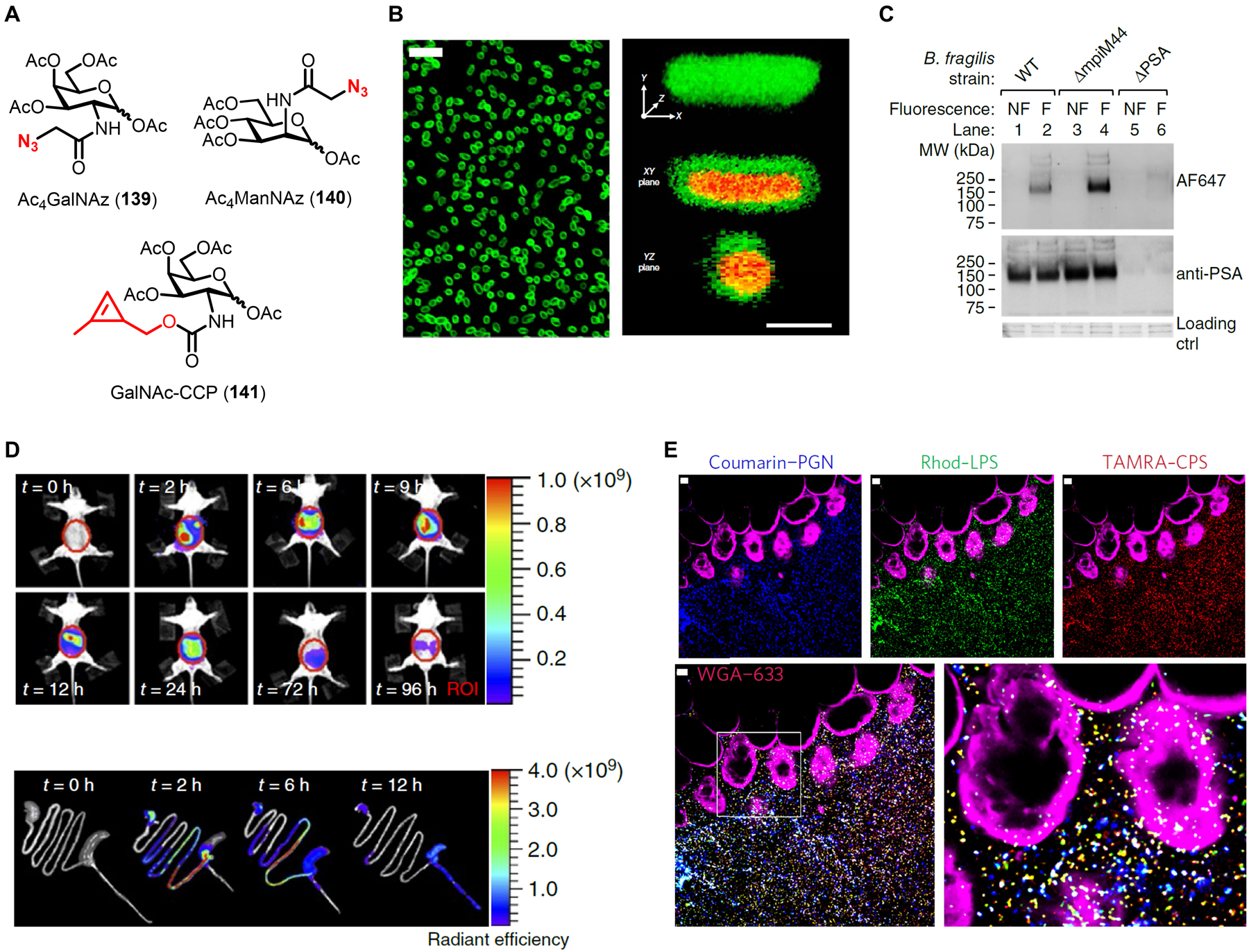
Similar articles
-
Development of Rare Bacterial Monosaccharide Analogs for Metabolic Glycan Labeling in Pathogenic Bacteria.ACS Chem Biol. 2016 Dec 16;11(12):3365-3373. doi: 10.1021/acschembio.6b00790. Epub 2016 Oct 28. ACS Chem Biol. 2016. PMID: 27766829 Free PMC article.
-
Self and nonself recognition with bacterial and animal glycans, surveys by synthetic chemistry.Methods Enzymol. 2010;478:323-42. doi: 10.1016/S0076-6879(10)78016-2. Methods Enzymol. 2010. PMID: 20816488
-
Synthesis and Application of Rare Deoxy Amino l-Sugar Analogues to Probe Glycans in Pathogenic Bacteria.ACS Infect Dis. 2022 Apr 8;8(4):889-900. doi: 10.1021/acsinfecdis.2c00060. Epub 2022 Mar 18. ACS Infect Dis. 2022. PMID: 35302355 Free PMC article.
-
Chemical biology tools to probe bacterial glycans.Curr Opin Chem Biol. 2024 Jun;80:102453. doi: 10.1016/j.cbpa.2024.102453. Epub 2024 Apr 5. Curr Opin Chem Biol. 2024. PMID: 38582017 Free PMC article. Review.
-
Tools for Studying Glycans: Recent Advances in Chemoenzymatic Glycan Labeling.ACS Chem Biol. 2017 Mar 17;12(3):611-621. doi: 10.1021/acschembio.6b01089. Epub 2017 Feb 13. ACS Chem Biol. 2017. PMID: 28301937 Free PMC article. Review.
Cited by
-
Chemical tools to study bacterial glycans: a tale from discovery of glycoproteins to disruption of their function.Isr J Chem. 2023 Feb;63(1-2):e202200050. doi: 10.1002/ijch.202200050. Epub 2022 Oct 18. Isr J Chem. 2023. PMID: 37324574 Free PMC article.
-
Selective Exoenzymatic Labeling of Lipooligosaccharides of Neisseria gonorrhoeae with α2,6-Sialoside Analogues.Chembiochem. 2022 Oct 6;23(19):e202200340. doi: 10.1002/cbic.202200340. Epub 2022 Aug 23. Chembiochem. 2022. PMID: 35877976 Free PMC article.
-
GLUT5: structure, functions, diseases and potential applications.Acta Biochim Biophys Sin (Shanghai). 2023 Oct 25;55(10):1519-1538. doi: 10.3724/abbs.2023158. Acta Biochim Biophys Sin (Shanghai). 2023. PMID: 37674366 Free PMC article. Review.
-
Chemical Proteomics Strategies for Analyzing Protein Lipidation Reveal the Bacterial O-Mycoloylome.J Am Chem Soc. 2024 May 1;146(17):12138-12154. doi: 10.1021/jacs.4c02278. Epub 2024 Apr 18. J Am Chem Soc. 2024. PMID: 38635392 Free PMC article.
-
A Bifunctional Chemical Reporter for in Situ Analysis of Cell Envelope Glycan Recycling in Mycobacteria.ACS Infect Dis. 2022 Nov 11;8(11):2223-2231. doi: 10.1021/acsinfecdis.2c00396. Epub 2022 Oct 26. ACS Infect Dis. 2022. PMID: 36288262 Free PMC article.
References
-
- Pikuta EV; Hoover RB; Tang J Microbial Extremophiles at the Limits of Life. Crit. Rev. Microbiol 2007, 33, 183–209. - PubMed
-
- Pai M; Behr MA; Dowdy D; Dheda K; Divangahi M; Boehme CC; Ginsberg A; Swaminathan S; Spigelman M; Getahun H et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076. - PubMed
-
- Sommer F; Bäckhed F The Gut Microbiota — Masters of Host Development and Physiology. Nat. Rev. Microbiol 2013, 11, 227–238. - PubMed
-
- Holst O; Moran AP; Brennan PJ Overview of the Glycosylated Components of the Bacterial Cell Envelope. In Microbial Glycobiology; Holst O; Brennan PJ; Itzstein M. v.; Moran AP, Eds.; Academic Press: San Diego, 2010, pp 1–13.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
