

RGEN-seq for highly sensitive amplification-free screen of off-target sites of gene editors
- PMID: 34880355
- PMCID: PMC8654851
- DOI: 10.1038/s41598-021-03160-8
RGEN-seq for highly sensitive amplification-free screen of off-target sites of gene editors
Abstract
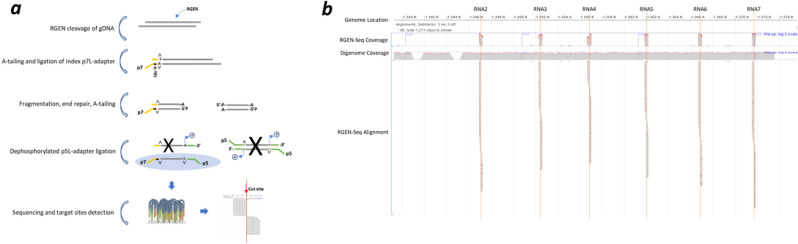
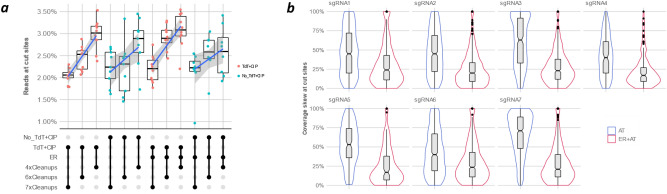
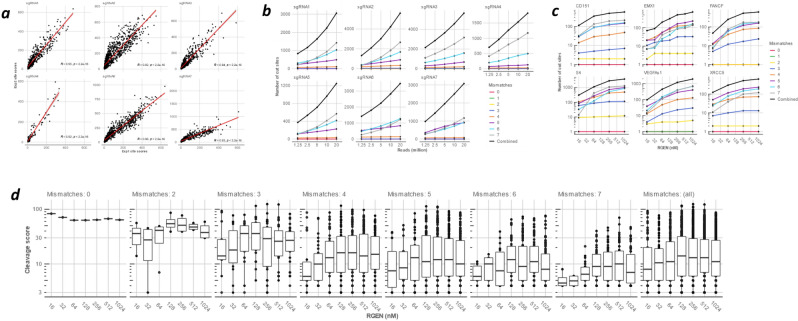
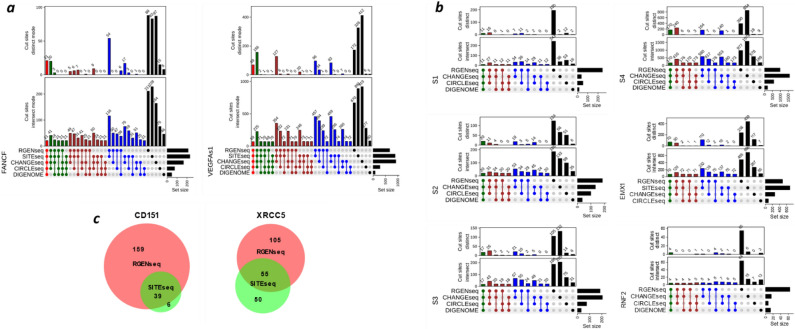
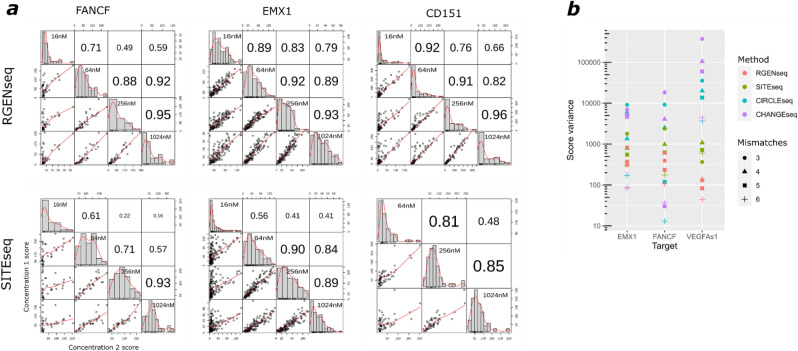
Sensitive detection of off-target sites produced by gene editing nucleases is crucial for developing reliable gene therapy platforms. Although several biochemical assays for the characterization of nuclease off-target effects have been recently published, significant technical and methodological issues still remain. Of note, existing methods rely on PCR amplification, tagging, and affinity purification which can introduce bias, contaminants, sample loss through handling, etc. Here we describe a sensitive, PCR-free next-generation sequencing method (RGEN-seq) for unbiased detection of double-stranded breaks generated by RNA-guided CRISPR-Cas9 endonuclease. Through use of novel sequencing adapters, the RGEN-Seq method saves time, simplifies workflow, and removes genomic coverage bias and gaps associated with PCR and/or other enrichment procedures. RGEN-seq is fully compatible with existing off-target detection software; moreover, the unbiased nature of RGEN-seq offers a robust foundation for relating assigned DNA cleavage scores to propensity for off-target mutations in cells. A detailed comparison of RGEN-seq with other off-target detection methods is provided using a previously characterized set of guide RNAs.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Identifying genome-wide off-target sites of CRISPR RNA-guided nucleases and deaminases with Digenome-seq.Nat Protoc. 2021 Feb;16(2):1170-1192. doi: 10.1038/s41596-020-00453-6. Epub 2021 Jan 18. Nat Protoc. 2021. PMID: 33462439
-
Tag-seq: a convenient and scalable method for genome-wide specificity assessment of CRISPR/Cas nucleases.Commun Biol. 2021 Jul 2;4(1):830. doi: 10.1038/s42003-021-02351-3. Commun Biol. 2021. PMID: 34215845 Free PMC article.
-
Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases.Genome Res. 2014 Jan;24(1):132-41. doi: 10.1101/gr.162339.113. Epub 2013 Nov 19. Genome Res. 2014. PMID: 24253446 Free PMC article.
-
Defining genome-wide CRISPR-Cas genome-editing nuclease activity with GUIDE-seq.Nat Protoc. 2021 Dec;16(12):5592-5615. doi: 10.1038/s41596-021-00626-x. Epub 2021 Nov 12. Nat Protoc. 2021. PMID: 34773119 Free PMC article. Review.
-
Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview.Int J Mol Sci. 2016 Sep 8;17(9):1507. doi: 10.3390/ijms17091507. Int J Mol Sci. 2016. PMID: 27618019 Free PMC article. Review.
Cited by
-
Regulatory Considerations for Genome-Edited T-cell Therapies.Cancer Immunol Res. 2024 Sep 3;12(9):1132-1135. doi: 10.1158/2326-6066.CIR-24-0482. Cancer Immunol Res. 2024. PMID: 39018097 Review.
References
-
- Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science346, (2014). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
