

Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy
- PMID: 34841295
- PMCID: PMC8607011
- DOI: 10.1016/j.xcrm.2021.100449
Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy
Abstract
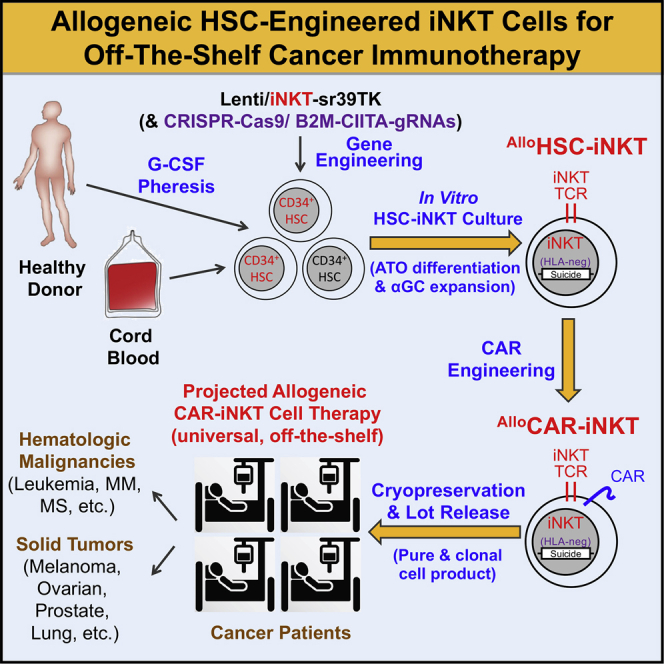
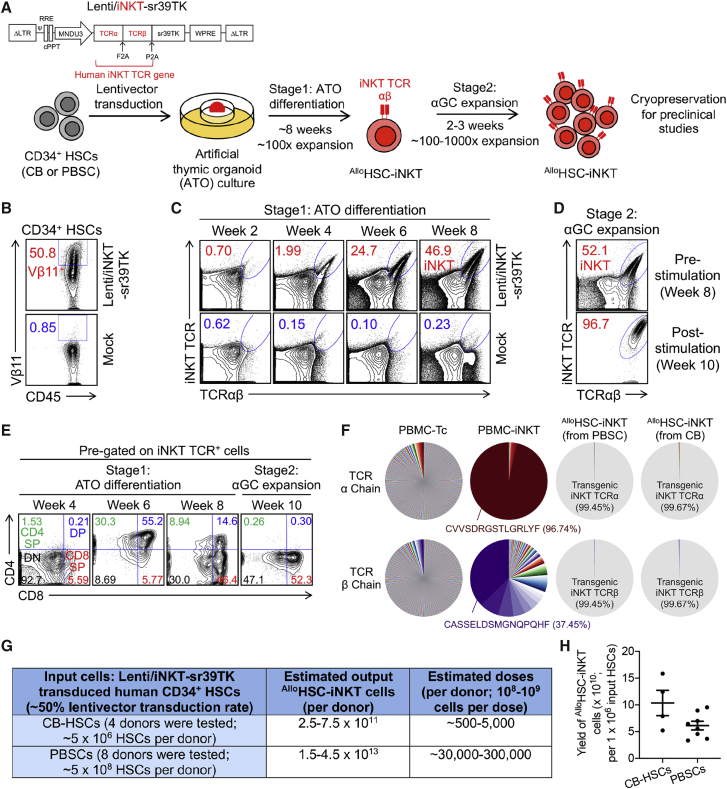
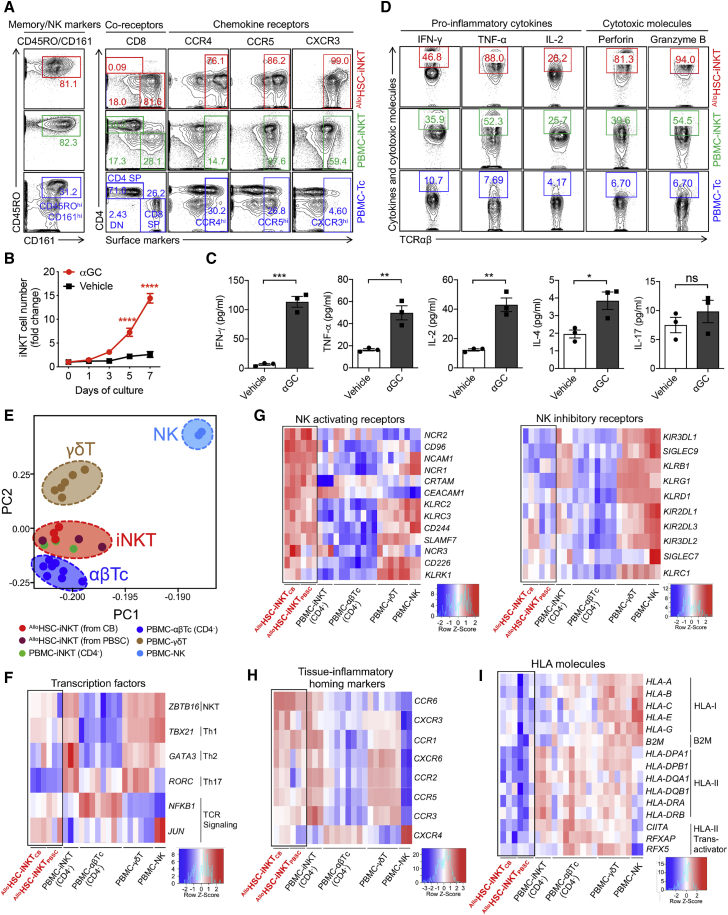
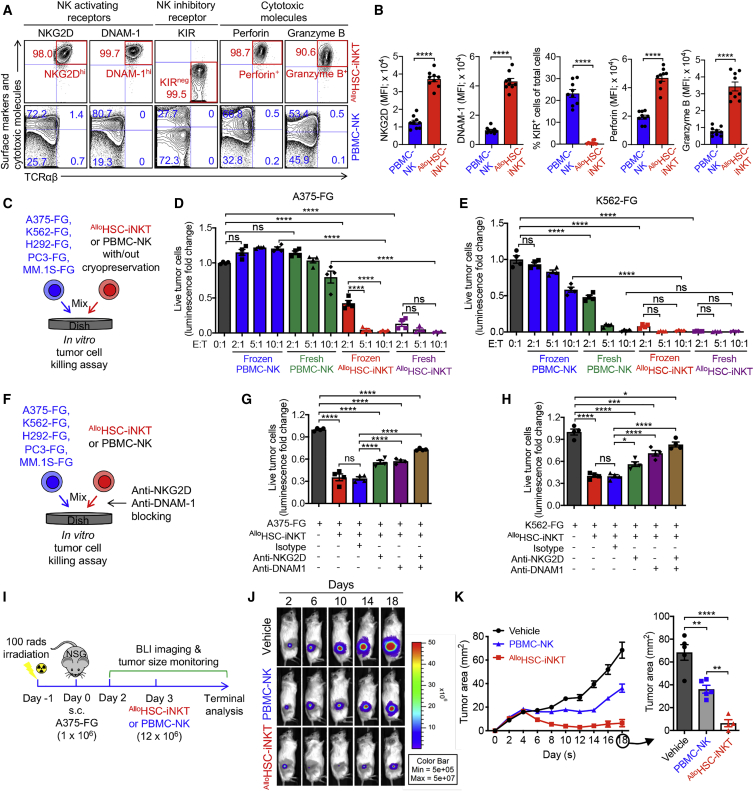
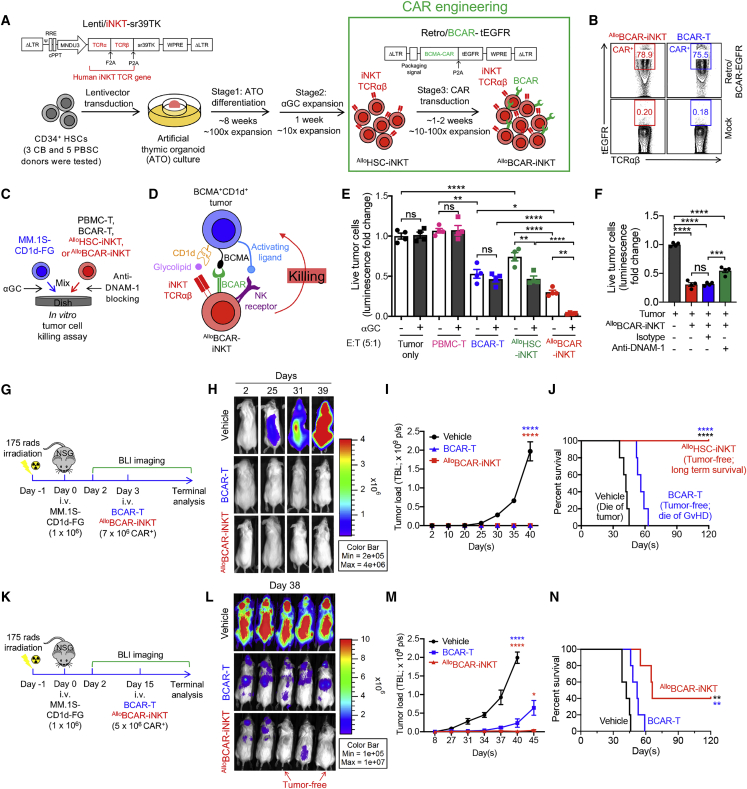
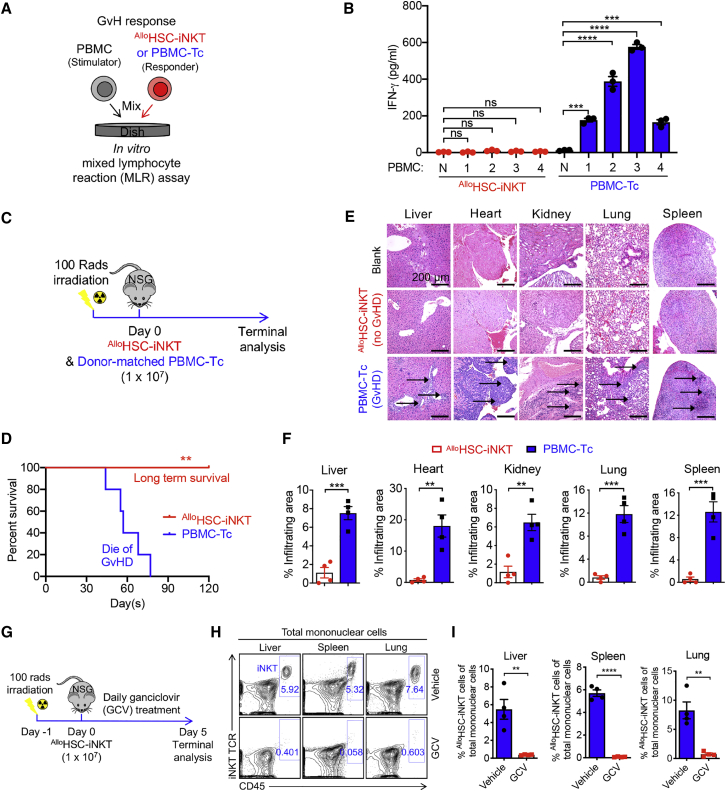
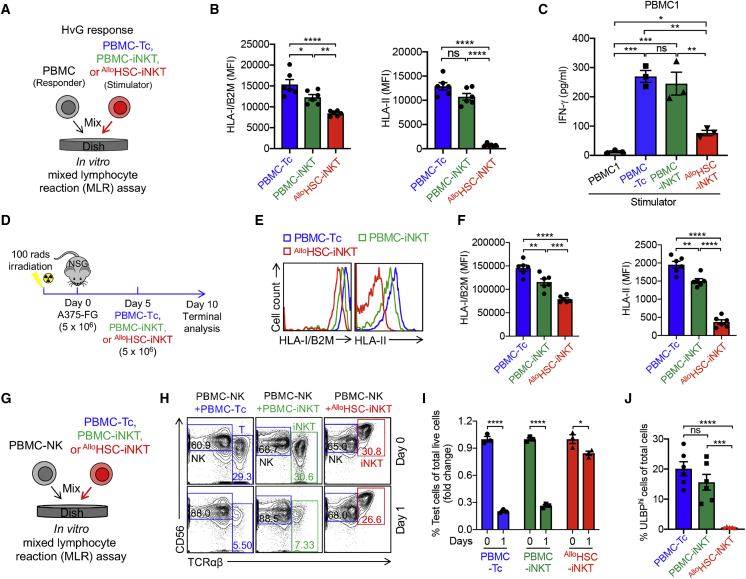
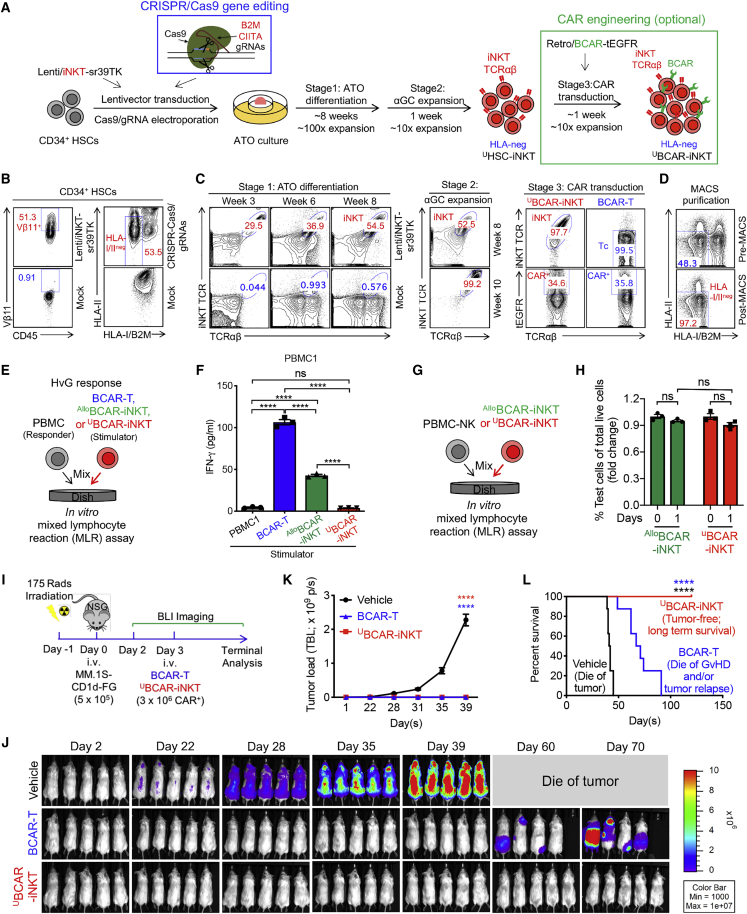
Cell-based immunotherapy has become the new-generation cancer medicine, and "off-the-shelf" cell products that can be manufactured at large scale and distributed readily to treat patients are necessary. Invariant natural killer T (iNKT) cells are ideal cell carriers for developing allogeneic cell therapy because they are powerful immune cells targeting cancers without graft-versus-host disease (GvHD) risk. However, healthy donor blood contains extremely low numbers of endogenous iNKT cells. Here, by combining hematopoietic stem cell (HSC) gene engineering and in vitro differentiation, we generate human allogeneic HSC-engineered iNKT (AlloHSC-iNKT) cells at high yield and purity; these cells closely resemble endogenous iNKT cells, effectively target tumor cells using multiple mechanisms, and exhibit high safety and low immunogenicity. These cells can be further engineered with chimeric antigen receptor (CAR) to enhance tumor targeting or/and gene edited to ablate surface human leukocyte antigen (HLA) molecules and further reduce immunogenicity. Collectively, these preclinical studies demonstrate the feasibility and cancer therapy potential of AlloHSC-iNKT cell products and lay a foundation for their translational and clinical development.
Keywords: CAR-engineered conventional αβ T cells; HLA-ablated universal HSC-iNKT cells; allogeneic HSC-engineered iNKT cells; allogeneic off-the-shelf cell therapy; allorejection; cancer immunotherapy; chimeric antigen receptor; graft-versus-host disease; hematopoietic stem cell; invariant natural killer T cells.
© 2021 The Authors.
Conflict of interest statement
Y.-R.L., Y.J.K., J.Y., P.W., Y. Zhu, G.M.C., A.M.-H., C.S.S., and L.Y. are inventors on patents relating to this study filed by UCLA. Y.J.K. is currently an employee of Nkarta. J.Y. and X.W. are currently employees of Appia Bio. X.C. is currently an employee of Atara Bio. Z.L. is currently an employee of Allogene. S.M.L. is a stockholder of 1200 Pharma and TORL BioTherapeutics. A.R. is a consultant for Amgen, Bristol-Meyers Squibb, Chugai, Genentech-Roche, Merck-MSD, Novartis, and Sanofi; a scientific advisory board member of and stockholder with Advaxis, Apricity, Arcus, Bioncotech, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Merus, Rgenix, and Appia Bio; and a co-founder and scientific advisory board member of Lutris, PACT Pharma, and Tango Therapeutics. J.S. is a consultant for Kite and on the speaker bureau for Kite and BMS. D.B.K. is a stockholder and scientific advisory board member of Allogene Therapeutics, Myogene Bio, ImmunoVec and Pluto Therapeutics. O.N.W. currently has consulting, equity, and/or board relationships with Trethera Corporation, Kronos Biosciences, Sofie Biosciences, Breakthrough Properties, Vida Ventures, Nammi Therapeutics, Two River, Iconovir, Appia BioSciences, Neogene Therapeutics, and Allogene Therapeutics. P.W. is a co-founder, stockholder, consultant, and advisory board member of HRain Biotechnology, TCRCure Biopharma, and Appia Bio. G.M.C., C.S.S., and A.M.-H. are cofounders and stockholders of Pluto Immunotherapeutics. L.Y. is a scientific advisor to AlzChem and Amberstone Biosciensec, and a co-founder, stockholder, and advisory board member of Appia Bio. None of the declared companies contributed to or directed any of the research reported in this article. The remaining authors declare no competing interests.
Figures
Similar articles
-
Engineering allorejection-resistant CAR-NKT cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy.Mol Ther. 2024 Jun 5;32(6):1849-1874. doi: 10.1016/j.ymthe.2024.04.005. Epub 2024 Apr 6. Mol Ther. 2024. PMID: 38584391
-
Development of off-the-shelf hematopoietic stem cell-engineered invariant natural killer T cells for COVID-19 therapeutic intervention.Stem Cell Res Ther. 2022 Mar 21;13(1):112. doi: 10.1186/s13287-022-02787-2. Stem Cell Res Ther. 2022. PMID: 35313965 Free PMC article.
-
Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer.Cell Stem Cell. 2019 Oct 3;25(4):542-557.e9. doi: 10.1016/j.stem.2019.08.004. Epub 2019 Sep 5. Cell Stem Cell. 2019. PMID: 31495780 Free PMC article.
-
Harnessing Chimeric Antigen Receptor-engineered Invariant Natural Killer T Cells: Therapeutic Strategies for Cancer and the Tumor Microenvironment.Curr Pharm Biotechnol. 2024;25(15):2001-2011. doi: 10.2174/0113892010265228231116073012. Curr Pharm Biotechnol. 2024. PMID: 38310449 Review.
-
Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies.Cells. 2021 Dec 10;10(12):3497. doi: 10.3390/cells10123497. Cells. 2021. PMID: 34944002 Free PMC article. Review.
Cited by
-
Advancing cell-based cancer immunotherapy through stem cell engineering.Cell Stem Cell. 2023 May 4;30(5):592-610. doi: 10.1016/j.stem.2023.02.009. Epub 2023 Mar 21. Cell Stem Cell. 2023. PMID: 36948187 Free PMC article. Review.
-
A milestone method to make natural killer T cells.Nat Biotechnol. 2024 May 14. doi: 10.1038/s41587-024-02243-x. Online ahead of print. Nat Biotechnol. 2024. PMID: 38744945 No abstract available.
-
Engineering allorejection-resistant CAR-NKT cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy.Mol Ther. 2024 Jun 5;32(6):1849-1874. doi: 10.1016/j.ymthe.2024.04.005. Epub 2024 Apr 6. Mol Ther. 2024. PMID: 38584391
-
Unlocking the potential of allogeneic Vδ2 T cells for ovarian cancer therapy through CD16 biomarker selection and CAR/IL-15 engineering.Nat Commun. 2023 Nov 8;14(1):6942. doi: 10.1038/s41467-023-42619-2. Nat Commun. 2023. PMID: 37938576 Free PMC article.
-
Advancements in adoptive CAR immune cell immunotherapy synergistically combined with multimodal approaches for tumor treatment.Bioact Mater. 2024 Sep 10;42:379-403. doi: 10.1016/j.bioactmat.2024.08.046. eCollection 2024 Dec. Bioact Mater. 2024. PMID: 39308543 Free PMC article. Review.
References
-
- Labanieh L., Majzner R.G., Mackall C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018;2:377–391. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
